







目前常用的去批次的软件有combat、combat_seq、removeBatchEffect。
芯片数据与处理过的非count数据可以使用SVA包的combat函数和limma包的removeBatchEffect函数。count数据(测序数据)使用SVA包的combat_seq函数。
combat函数是基于贝叶斯框架下的调整数据批次效应的函数,主要适用于已经被过滤和标准化后的数据(芯片数据/非count数据)。
ComBat-seq使用负二项回归模型来去除批次效应,通过将原始数据映射到预期分布来提供调整后的数据。
rm(list=ls())
setwd("/工作安排/去批次处理数据/")
## 1. 载入有批次效应的数据
info <- read.csv("info.csv",row.names = 1)
count <- read.csv("count-merge.csv",row.names = 1)
info[,2] <- as.factor(info$type)##type表示的是生物学上的不同处理
info[,3] <- as.factor(info$batch)##batch表示不同的批次信息
row.names(info) <- info$sample
exp=na.omit(count) #去除缺失值
## 2. 不做校正的PCA分析
#install.packages("FactoMineR")
#install.packages("factoextra")
library(FactoMineR)
library(factoextra)
pre.pca <- PCA(t(exp),graph = FALSE)
fviz_pca_ind(pre.pca,
geom= "point",
col.ind = info$batch,
addEllipses = TRUE,
legend.title="Group" )
## 3. Sva包combat消除批次效应
#建立批次效应的模型,info$type表示的是数据中除了有不同的批次,还有生物学上的差异。
#在校正的时候要保留生物学上的差异,不能矫正过枉。
model <- model.matrix(~as.factor(info$type))
##消除批次效应
#if (!require("BiocManager", quietly = TRUE))
# install.packages("BiocManager")
#BiocManager::install("sva") ##sva依赖于4.4版本R包
library(sva)
combat_Expr <- ComBat(dat = exp,batch = info$batch,mod = model)
##combat_Expr就是校正后的数据,有负值,后续使用limma分析
write.csv(combat_Expr, file = './count-merge-combat.csv')
## 校正后的PCA分析
library(FactoMineR)
combat.pca <- PCA(t(combat_Expr),graph = FALSE)
fviz_pca_ind(combat.pca,
geom= "point",
col.ind = info$batch,
addEllipses = TRUE,
legend.title="Group" )
# 4.Sva包combat_Seq消除批次效应combat_seq。专门针对 RNA-Seq 计数(即count矩阵)数据
expr_count_combat <- ComBat_seq(counts = as.matrix(exp),
batch = info$batch,
group = info$type)
write.csv(expr_count_combat, file = './count-merge-combatSeq.csv')
## 校正后的PCA分析
library(FactoMineR)
aft.pca <- PCA(t(expr_count_combat),graph = FALSE)
fviz_pca_ind(aft.pca,
geom= "point",
col.ind = info$batch,
addEllipses = TRUE,
legend.title="Group" )
## 5. removeBatchEffect去批次
library(limma)
mod <- model.matrix(~factor(coldata$type))
expr_limma <- removeBatchEffect(exp, batch = info$batch, design=mod)
## 校正后的PCA分析
aft.pca <- PCA(t(expr_limma),graph = FALSE)
fviz_pca_ind(aft.pca,
geom= "point",
col.ind = info$batch,
addEllipses = TRUE,
legend.title="Group" )
批次效应不能完全消除,只是尽可能的减少了批次带来的干扰!去批次之后若出现负值,后续使用limma进行差异分析。
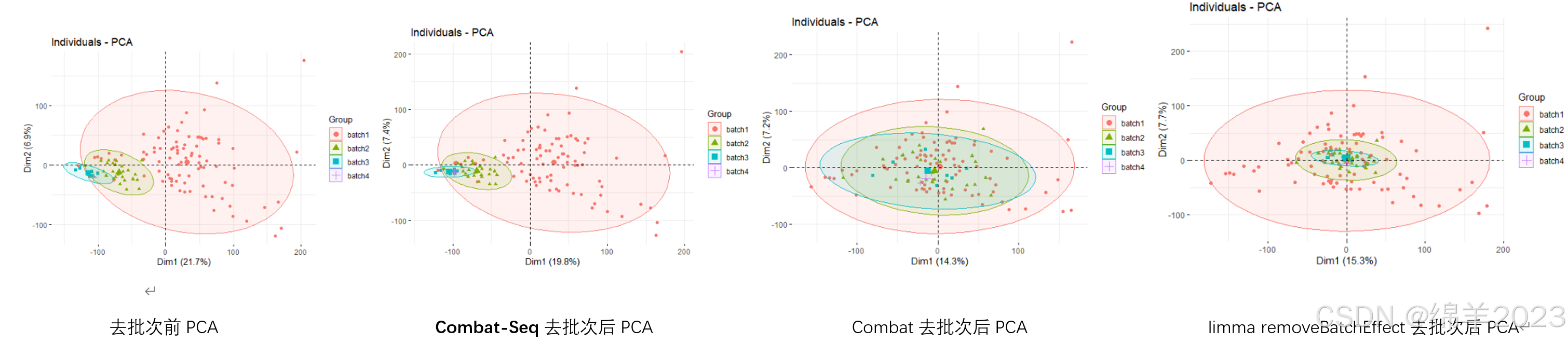
本次count数据使用combat-Seq去批次效果不明显,可能原因:数据之间本身批次小;batch1样本大,本身差异大。
同时使用其他工具进行去批次,发现效果较好,有人使用combat和removeBatchEffect对count数据进行去批次,可行性有待商榷。
欢迎大家讨论与指正不足。
参考
批次效应去除之combat和removebatcheffect_combat去批次-CSDN博客
转录组数据去批次方法整理(combat,combat-seq,removeBatchEffect)_去除批次效应-CSDN博客

















 3275
3275

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









