







 本文介绍了DeepMind的AlphaFold模型在蛋白质结构预测上的应用,通过特征工程、深度学习和结构生成步骤,解决蛋白质折叠问题。作者复现了预测过程,指出该模型对CPU和GPU计算的需求,并分享了训练与预处理的实践经验。
本文介绍了DeepMind的AlphaFold模型在蛋白质结构预测上的应用,通过特征工程、深度学习和结构生成步骤,解决蛋白质折叠问题。作者复现了预测过程,指出该模型对CPU和GPU计算的需求,并分享了训练与预处理的实践经验。
实验|Vachel 算力支持|幻方AIHPC

要说去年一年AI领域的最重大进展,DeepMind在Nature上发表的“AlphaFold”无出其右,其旨在根据一个蛋白质的氨基酸序列来确定它的3D结构,是生物学领域中的几个重大挑战课题之一。在“蛋白质结构预测关键评估”(CASP)的蛋白质结构预测年赛上,AlphaFold击败了其他百来支团队并取得了惊人的效果,有些时候,AlphaFold预测的结构与利用X射线晶体学和近几年的冷冻电镜(cryo-EM)等“金标准”实验方法所确定的结构几乎一样。科学家说,目前看来,AlphaFold暂时还不能取代这些费力又昂贵的实验方法,但它将带来全新的研究生命的方式。
作为一个计算机专业的算法从业人员,本期笔者带着浓厚的兴趣来尝试一下DeepMind开源的Alphafold模型并验证效果。我们复现了Alphafold的训练与预测过程,为大家带来第一手的测试体验。
模型介绍
AlphaFold解决的问题是蛋白质折叠问题。输入是一个氨基酸序列,每一个位置代表一个元素,输出是一个拓扑结构,如下图所示:
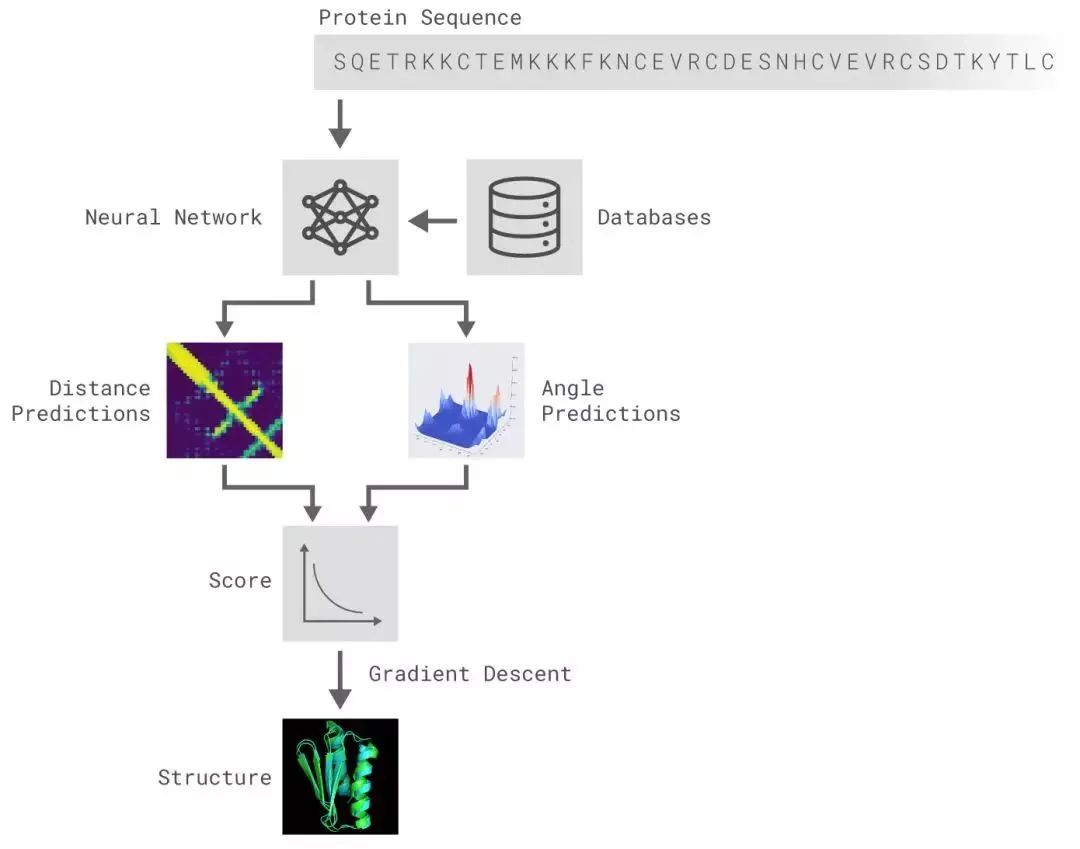
直接输出一个拓扑结构对于深度学习来说比较困难,一般需要输出结构化的数据。DeepMind将该问题转化为预测氨基酸链的一些性质。准确的来说,深度模型输出的是每一个氨基酸单元和其下一个氨基酸单元在空间中的夹角与距离,再组合成拓扑结构。
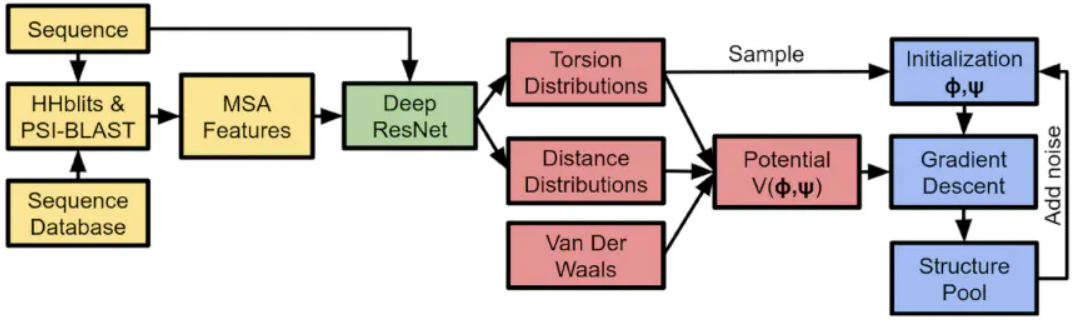
如上图所示,AlphaFold算法大致分为以下几个部分:
-
特征工程:序列和MSA特征抽取,结合专家经验数据库,把氨基酸链的输入转换到特征空间;
-
深度神经网络结构预测:依据1中的特征预测氨基酸链的一些性质,比如氨基酸之间两两的距离分布,氨基酸链的夹角分布;
-
Potential Construction:结合专家经验构造一个评估函数,来评估2中神经网络输出解的合理程度;
-
结构生成:对于2中预测的距离分布、夹角分布,使用3中的评估函数评估Loss,然后使用梯度下降法优化,直到收敛。
可以看到
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章



















 1509
1509

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











