







根据前三节的学习,已经获得了矩阵、分组信息等,接下来就可以做差异分析。本节课用的是limma包,也可以选择其他包,参考文章:用limma对芯片数据做差异分析。
使用说明
使用这个包需要三个数据:
- 表达矩阵 exprSet
- 分组矩阵 design
- 差异比较矩阵
三个步骤:
- lmFit 线性拟合
- eBayes 贝叶斯检验
- topTable 生成结果
1.差异分析
表达矩阵
表达矩阵就是前三节最后处理得到的exprSet,行名是探针转换得到的基因名,列名是对照组和处理组。
分组矩阵
就是展示每一个样品属于对照还是处理,1代表是,0代表不是。
> dim(exprSet)
[1] 18851 6
> library(limma)
#告诉design 哪几个样品是对照 哪几个样品是处理 1代表是 0代表不是
#查看group_list和factor(group_list) 明确自己在做什么
> group_list
[1] "control" "control" "control" "vemurafenib"
[5] "vemurafenib" "vemurafenib"
> factor(group_list)
[1] control control control vemurafenib vemurafenib
[6] vemurafenib
Levels: control vemurafenib
> design<-model.matrix(~0+factor(group_list))
> colnames(design)=levels(factor(group_list))
> rownames(design)=paste(group_list,1:3,sep='')
> design
control vemurafenib
control1 1 0
control2 1 0
control3 1 0
vemurafenib1 0 1
vemurafenib2 0 1
vemurafenib3 0 1
attr(,"assign")
[1] 1 1
attr(,"contrasts")
attr(,"contrasts")$`factor(group_list)`
[1] "contr.treatment"
比较矩阵
把vemurafenib组跟control组进行差异分析比较,不能弄反。
#做比较矩阵 Vemurafenib和control比较
> contrast_matrix<-makeContrasts(vemurafenib-control,levels = design)
> contrast_matrix
Contrasts
Levels vemurafenib - control
control -1
vemurafenib 1
差异分析
参考文章:r<-差异分析】当使用limma时,它在比较什么。
#差异分析
> library(limma)
#ImFit 线性模型拟合
> fit<-lmFit(exprSet,design)
#根据对比模型进行差值计算
> fit2<-contrasts.fit(fit,contrast_matrix)
#eBayes 贝叶斯检验
> fit2<-eBayes(fit)
#生成所有基因的检验结果报告
> temOutput=topTable(fit2,coef = 1,n=Inf)
> nrDEG=na.omit(temOutput)
> head(nrDEG)
logFC AveExpr t P.Value adj.P.Val B
HSPA8 14.18073 13.95993 303.5779 4.964686e-23 2.315522e-20 39.49335
H1-4 14.06587 13.95178 300.1022 5.638639e-23 2.315522e-20 39.44253
GAPDH 14.34823 14.35550 299.4269 5.780821e-23 2.315522e-20 39.43250
RPLP0 13.99060 14.04047 296.7633 6.380983e-23 2.315522e-20 39.39243
RPL18 13.98733 13.99132 295.4763 6.695019e-23 2.315522e-20 39.37278
RACK1 14.02343 14.05142 295.4697 6.696674e-23 2.315522e-20 39.37268
检查差异矩阵是否有误
由表达矩阵可以知道基因CD36在各个组的表达情况,对照组大概是4,处理组大概是10,logFC(FC=foldchange即处理比对照)应该是正数,从nrDEG矩阵里可以查看证实logFC确实是正值,则说明没有问题。
#检查差异分析矩阵是否有误 以CD36为例
> exprSet[rownames(exprSet)=="CD36",]
control1 control2 control3 vemurafenib1 vemurafenib2 vemurafenib3
4.54610 4.40210 4.49239 10.25060 10.21480 10.31570
> nrDEG[rownames(nrDEG)=="CD36",]
logFC AveExpr t P.Value adj.P.Val B
CD36 4.480197 7.370282 87.49708 4.634617e-17 5.22656e-17 29.78431
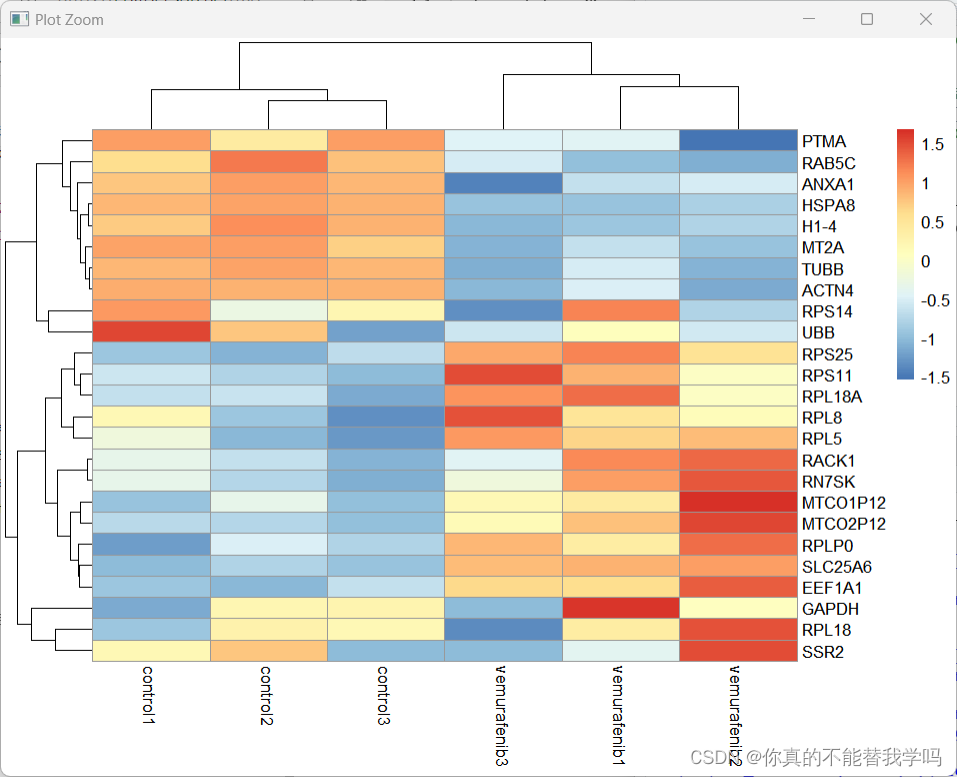
热图
#热图 只取前25个
library(pheatmap)
getgene=head(rownames(dif),25) #前25个名字
getgene.matrix=exprSet[getgene,]
getgene.matrix=t(scale(t(getgene.matrix)))
pheatmap(getgene.matrix)
scale()将一组数进行处理,默认情况下是将一组数的每个数都减去这组数的平均值后再除以这组数的均方根。
2.火山图
EnhancedVolcano(nrDEG,
lab=rownames(nrDEG),
x='logFC',
y='P.Value',
xlim=c(-6,6),
title='Vemurafenib versus control',
pCutoff=0.01,
FCcutoff=2.0,
col=c('black','blue','green','red1'),
colAlpha=1)
图有bug,还不知道问题在哪。
3.GO KEGG GSEA分析
3.1功能富集分析介绍
定义:
在某一特定基因子集中,多个基因共同参与的pathway通路,与标准背景相比,是否比例升高、具有统计显著性。
原理:
富集的统计学基础是超几何分布,简单来说根据下面的Fisher精确检验(Fisher exact test)公式,对每个GO或KEGG term计算一个p值:
p=(M/K)[(N-M)/(n-k)]/(N/n),其中
N:所有gene总数
n:N中差异表达gene的总数
M:N中属于某个GO term的gene个数
k: n中属于某个GO term的gene个数
p:表示差异表达gene富集到这个GO term上的可信程度
当p<0.05或0.01,则认为差异表达gene显著到这个GO term上(自己定义p值),表明提供的信息更集中,更有意义。
富集分析类别:
- 差异基因富集分析:不需要表达值 只需要gene name 【GO KEGG】
- 基因集(gene set)富集分析 不管有无差异 需要全部genes表达值【GSEA】
基因集富集分析的好处:可以发现被舍弃的genes可能参与了某重要生理过程或信号通路,工具GSEA。
富集什么:
最常用的基因注释工具是GO和KEGG,这基本上是差异基因分析一定做的两件事情。
GO可以在BP(生物过程)、MF(分子功能)、CC(细胞组分) 三个方面进行注释,较多的是BP(生物过程)。
KEGG将基因组信息和高一级的功能信息有机地结合起来,通过对细胞内已知生物学过程的计算机化处理和将现有功能解释标准化,对基因的功能进行系统化的分析。另一个任务是将基因组的一系列基因用一个细胞内的分子相互作用的网络连接起来的过程,如一个通路或是有个复合物,通过他们来展现更高一级的生物学功能。
KEGG物种缩写
生信技能树使用教程
GSEA(Gene Set Enrichment Analysis)的基本思想是使用预定义的基因集(通常来自功能注释或先前实验的结果),将基因按照在两类样本中的差异表达程度排序,然后检验预先设定的基因集合是否在这个排序表的顶端或者底端富集。基因集合富集分析检测基因集合而不是单个基因的表达变化,因此可以包含这些细微的表达变化,预期得到更为理想的实验结果。
3.2GSEA
概念:
GSEA即基因集富集分析,集就是某一个通路(相关的所有的基因的总和),专业说法:在以前的实验中发表的数据或表达谱上共表达的基因信息数据集合。
GSEA分析的是什么:
我们在利用DESeq2、edgeR或者Llimma进行差异分析笔尖后,会得到一个列表,里面有所有的差异基因。对这个列表里的基因表达水平从高到低进行排序,GSEA分析就是确定一个基因集S(或一个通路)的成员是否倾向于出现在列表L的顶部或者底部,如果聚集在顶部/底部,该基因集与表型分类区分相关。GSEA是一种无阈值方法,可根据其差异表达等级或其他分数对所有基因进行分析,无需事先进行基因过滤。
原理:
通俗即是:假设某一个通路的全部基因在排序后的差异基因列表中随机分布,但是如果我们看到他们出现在基因列表的某一侧(从图上看就是在某一侧形成一个峰),那么就可以计算显著性来看富集程度如何,如果富集结果显著,那么就拒绝原假设,认为这个通路的基因在我们的基因列表中富集,并且可以看到富集分数。
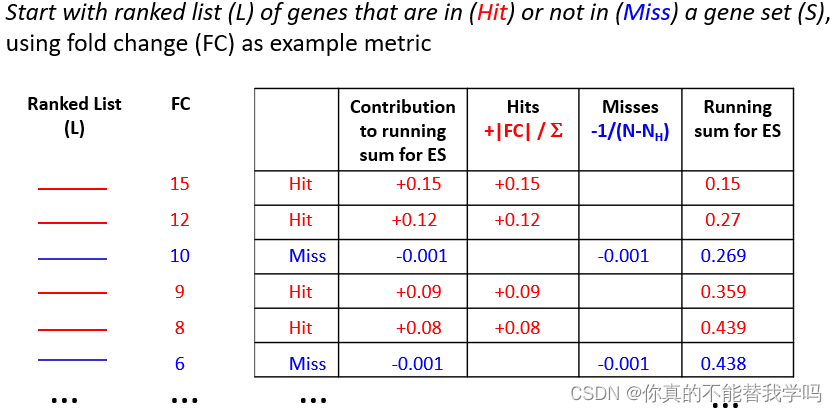
富集分数:
Enrichment Score,简称ES,从我们排序的差异分析列表里,从高到低一个一个看,遇到一个基因,如果属于我们要的基因集S,ES就会加分,反之就会减分。每一步统计值增加或减少的幅度与基因的表达变化程度(更严格的是与基因和表型的关联度)是相关的,富集得分ES最后定义为最大的峰值,正值ES表示基因集在列表的顶部富集,负值ES表示基因集在列表的底部富集。
图中Hit是属于,Miss不属于。
实际操作:R















 3752
3752











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









