






目录
1.1 UPGMA(Unweighted pair group method with arithmetic mean,平均连接聚类法)
1.2 ME(Minimum Evolution,最小进化法)
1.3 NJ(Neighbor-Joining,邻接法)编辑
3、在重建进化树过程中,均需选择bootstrap进行树的检验
构建进化树的算法
1、基于距离
1.1 UPGMA(Unweighted pair group method with arithmetic mean,平均连接聚类法)
- 基本假设:该方法假设在进化过程中,每一世系发生趋异的次数相同,即核苷酸或氨基酸的替换速率是均等且恒定的。
- 流程:

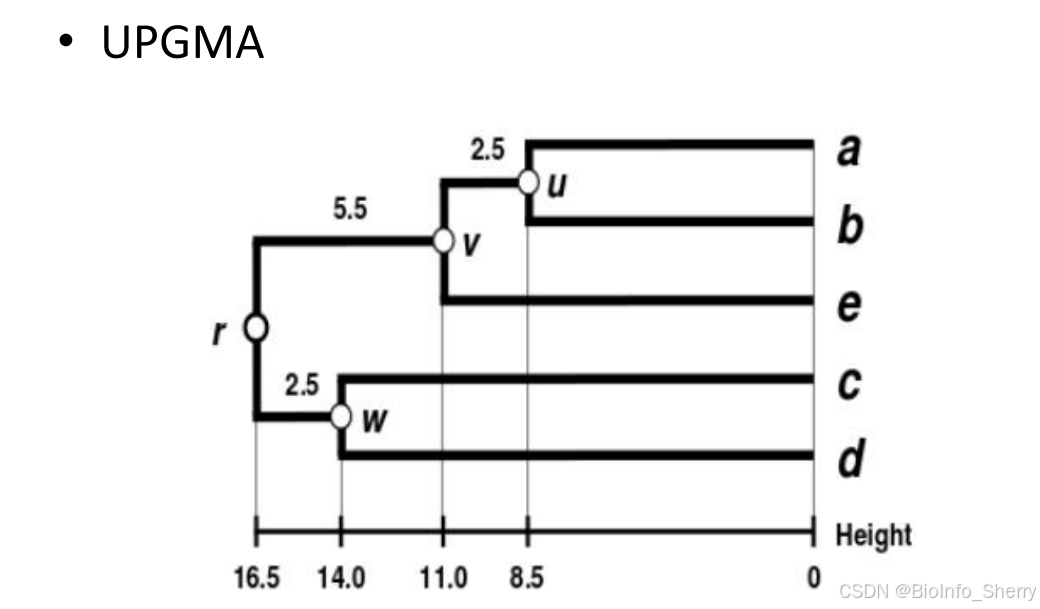
- 优点在于计算效率高,适合快速构建初步的系统发育树,但由于其假设条件较为严格,可能不适用于所有类型的数据集
1.2 ME(Minimum Evolution,最小进化法)
1.3 NJ(Neighbor-Joining,邻接法)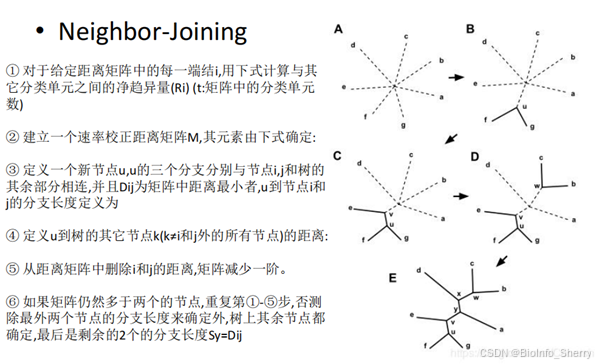
2、基于特征
2.1 最大简约法(Maximum Parsimony)
- 理论基础:奥卡姆剃刀
- 方法:计算所有可能的拓扑结构;计算出所需替代数最小的那个拓扑结构,作为最优树。
- 特点:用于分析如插入、缺失等序列。在分析序列上存在较多的回复突变或平行突变,而被检验的序列位点数又比较少的时候,最大简约法可能会给出一个不合理的或者错误的进化树推导结果。
2.2 最大似然法(Maximum Likelyhood)
- 原理:对所有可能的系统发育树都计算似然函数,选择似然函数值最大的为最优树。
- 需要先确定序列进化的模型,如Jukes—Cantor模型、Kimura二参数模型及一般二参数模型等。
- 优点:在进化模型选择合理的情况下,ML法与进化事实吻合最好。缺点:计算强度非常大,极为耗时。

- 计算
- P矩阵(Probability Matrix)也称为转移概率矩阵,描述了在给定分支长度上从一个状态转移到另一个状态的概率【e.g.PAM矩阵】;特性:无记忆,可逆。
- Q矩阵(Instantaneous Rate Matrix)也称为瞬时速率矩阵,包含了核苷酸(或氨基酸)之间替换的瞬时速率。Q矩阵中的每个元素Qij表示单位时间内从核苷酸(或氨基酸)i转变为核苷酸(或氨基酸)j的速率。

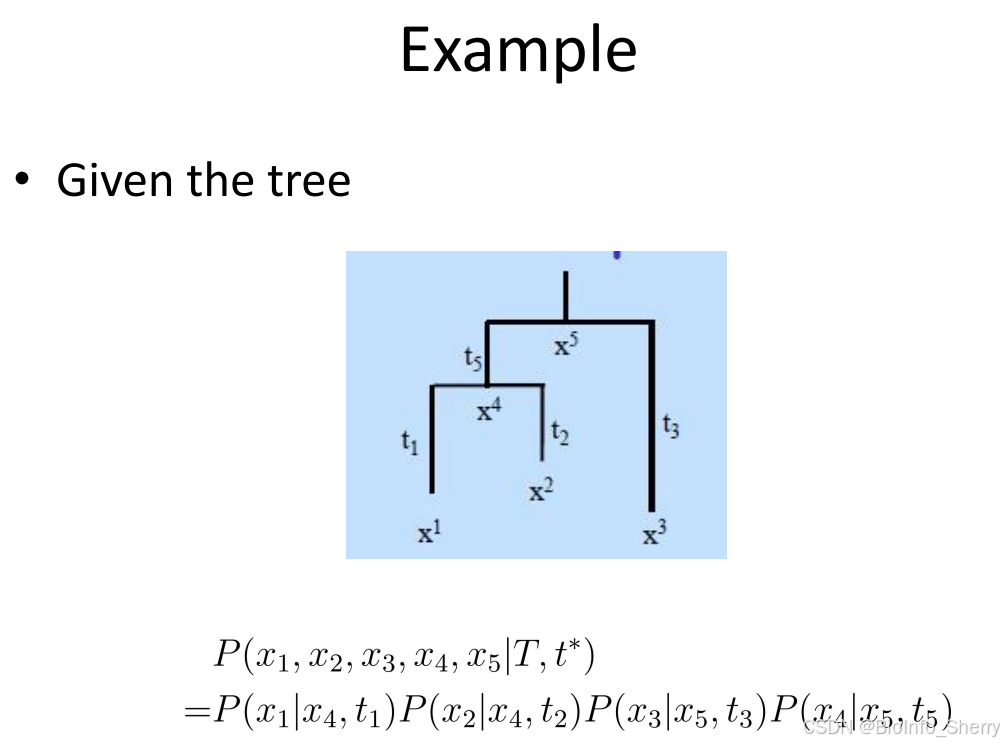
- 计算未对齐的可能性


2.3 进化简约法(EP法)
2.4 相容性方法
算法的对比与选择
1、算法选择
如果序列的相似性较高,各种方法都会得到不错的结果,模型间的差别也不大
若有合适的分子进化模型可供选择,用最大似然法构树获得的结果较好
对于近缘物种序列,通常情况下使用最大简约法
而对于远缘物种序列,一般使用邻接法或最大似然法
对于相似度很低的序列,邻接法往往出现长枝吸引(branch attraction)现象,有时严重干扰进化树的构建
2、邻接法和最大似然法需要选择模型
蛋白质序列的构树模型一般选择Poisson correction(泊松修正)
核酸序列的构树模型一般选择Kimura 2-parameter (Kimura-2参数).
3、在重建进化树过程中,均需选择bootstrap进行树的检验
• 一般bootstrap的值>70,则认为重建的进化树较为可靠。
• 如果bootstrap的值太低,则有可能进化树的拓扑结构有错误,进化树是不可靠的。
• 一般推荐用两种以上不同的方法构建进化树,如果所得到的进化树类似,且 bootstrap值总体较高,则得到的结果较为可靠。
4、软件与平台

考试重点
主要是理解概念,集中在UPGMA、NJ、最大简约法、最大似然法

















 6268
6268

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









