






RNA测序(RNA-seq)已成为现代生物学和临床科学中的典范技术,其广泛应用得益于生物信息学界不断开发的计算工具。RNA-seq可用于检测新的外显子、评估基因表达和研究可变剪接结构,但原始数据存在规模大、测序技术有局限等问题,需要多种计算工具进行分析。本文概述了RNA测序(RNA-seq)数据分析的完整流程,从原始数据到生物学解读。文章强调了数据质量控制、预处理、差异表达分析和功能注释的重要性,并介绍了常用的生物信息学工具和方法,助力研究人员合理运用现有计算工具。
 标题:RNA-seq data science: From raw data to effective interpretation
标题:RNA-seq data science: From raw data to effective interpretation
网址:
https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2023.997383/full
RNA测序
RNA-seq利用核酸高通量测序确定RNA分子的核苷酸序列和特定RNA种类的数量,其分析需借助专门计算工具处理测序技术的缺陷。RNA-seq文库制备始于从生物样本中提取和分离RNA,对于短读长测序,后续还需逆转录、PCR扩增和片段化处理,最终由测序平台生成读段。
1. 实验设计要点
• 样本选择:生物学重复≥3,避免个体差异干扰
• 文库构建:选择合适片段大小(mRNA:200-500bp)
• 质量控制:使用Agilent Bioanalyzer检测完整性(RIN>7)
2. 高通量RNA-seq技术
RNA-seq通过逆转录构建cDNA文库,利用高通量测序技术获取数百万条RNA片段序列。
 图1 Overview of RNA-seq
图1 Overview of RNA-seq
常用的高通量RNA-seq技术平台有Illumina、Nanopore和PacBio。Illumina测序基于边合成边测序化学原理,于2006年商业化,能并行测定大量cDNA片段序列,测序通量高;Nanopore测序2014年推出,属于第三代测序技术,可直接对任意长度的天然DNA和RNA片段测序,无需化学修饰或PCR扩增;PacBio测序即单分子实时(SMRT)测序,2010年问世,可生成全长cDNA序列,长读段在单分子尺度准确性高,但受样本长度限制。这些技术各有优劣,短读长技术错误率低、通量高,但转录组重建和定量困难;长读长测序能提升组装准确性、获取更多转录本信息,但错误率高、通量低。混合方法可综合两者优势,但成本和材料需求更高。
RNA-seq数据科学:从原始数据到有效解读
RNA-seq可用于揭示疾病相关的基因和蛋白问题,计算分析是解读生物转录组复杂性的关键。下面将介绍RNA-seq数据分析的主要步骤,从原始数据处理到获得生物学见解。
数据获取
RNA-seq数据通常以FASTQ格式存储,包含测序读段和相应的质量分数。数据可以从公共数据库如NCBI的SRA(Sequence Read Archive)获取。
原始数据质量控制
质量控制是RNA-seq数据分析的第一步。常用工具包括FastQC和MultiQC,用于评估读段质量、GC含量和序列重复性。低质量读段可以使用Trimmomatic或Cutadapt等进行修剪。
读段比对
读段比对是RNA-seq分析的重要环节,通过与注释的参考转录组比对,可确定读段在基因组中的来源和覆盖度。但该方法难以发现参考序列中缺失的转录本。常用比对工具包括Star、TopHat等,部分工具如GenomeScope、Smudgeplot和Merqury能在不映射读段的情况下估计覆盖度。当读段比对到多个转录本时,MapSplice、HISAT2等软件可通过正确比对跨越外显子 - 内含子接头的读段,减少多映射问题,为参考引导的组装奠定基础。若读段与参考序列存在错配,可借助Pegasus等工具,通过de novo assembly、识别跨越融合接头的读段等策略,区分测序错误和生物学变异,并进行基因识别和分类。
基因表达定量分析
RNA-seq可在可变转录本水平对基因表达进行定量分析,包括转录本和基因表达量的估计以及差异表达分析。
 图2 可变剪接与RNA测序技术
:遗传信息从DNA开始,DNA由内含子和外显子组成,转录成前体mRNA后经剪接形成成熟mRNA并翻译为蛋白质。可变剪接或外显子跳跃可产生不同的转录本。RNA-seq读段是从转录本中采样的短序列,可由Illumina(产生短读段)、Nanopore和PacBio(产生长读段)等平台生成。图中展示了唯一比对的读段分别与参考转录组和参考基因组比对的两种情况,部分读段跨越外显子 - 外显子接头,而一些较短的读段仅比对到单个外显子。TSS为转录起始位点;TES为转录终止位点。
图2 可变剪接与RNA测序技术
:遗传信息从DNA开始,DNA由内含子和外显子组成,转录成前体mRNA后经剪接形成成熟mRNA并翻译为蛋白质。可变剪接或外显子跳跃可产生不同的转录本。RNA-seq读段是从转录本中采样的短序列,可由Illumina(产生短读段)、Nanopore和PacBio(产生长读段)等平台生成。图中展示了唯一比对的读段分别与参考转录组和参考基因组比对的两种情况,部分读段跨越外显子 - 外显子接头,而一些较短的读段仅比对到单个外显子。TSS为转录起始位点;TES为转录终止位点。
1.转录本和基因表达的估计
计算方法通过统计与单个参考转录本匹配的读段数量来估计基因和转录本的表达水平。HT-Seq-count、Rcount和featureCounts等工具应用广泛,但基于计数的方法在处理短读段时,难以准确估计不同异构体的表达水平。一种保守方法是仅考虑能唯一比对到单个转录本的读段,另一种方法则是通过概率分配将读段映射到可能的异构体上。此外,Pseudoalignment方法如Sailfish、Salmon和Kallisto,利用预编译的k-mer库,可实现快速定量,且准确性与传统比对方法相当。定量结果通常以计数矩阵形式存储。
2.差异基因表达分析
在估计基因和转录本表达水平后,需运用统计方法检测不同实验条件下的表达差异。差异表达分析旨在识别在不同条件下表达显著变化的基因。常用工具包括DESeq2、edgeR和limma。这些工具基于统计模型,考虑测序深度和生物学变异。由于实验样本的相对性和mRNA水平的动态变化,需要进行统计检验。常用的p值可判断基因表达差异的显著性,一般以p值小于0.05为差异显著,但该阈值可根据数据噪声调整。统计检验存在I型错误和II型错误,可通过控制家族错误率或错误发现率来调整p值,以减少假阳性结果。
在进行差异表达分析时,还需考虑噪声因素,如批次效应和生物学批次效应,现有统计方法可有效检测和调整这些隐藏的混杂因素。此外,还有其他分析方法,如最小显著差异法、广义线性模型(GLM)框架和概率正对数比(PPLR)等,可提供更准确的结果。根据RNA-seq数据的归一化类型,还可使用机器学习方法,如基于Poisson或负二项分布的分类模型,或通过Bioconductor MLSeq软件包进行分析,以识别差异表达基因。差异表达分析还可结合表达数量性状位点(eQTL)分析,其检测能力受样本测序深度、基因次要等位基因频率等因素影响,常用变换和线性回归模型进行检测。最后,差异表达分析结果可通过定量PCR(qPCR)等独立技术进行验证。
差异表达结果可以通过火山图、热图和MA图进行可视化。常用工具包括ggplot2和pheatmap。
等位基因特异性表达的测量
RNA-seq可测量等位基因特异性表达(ASE),以揭示遗传变异的顺式调控效应。在典型的RNA-seq实验中,只有在基因转录区域包含杂合单核苷酸多态性(aseSNP)时才能测量ASE,aseSNP可用于识别来自每个基因拷贝的读段。等位基因不平衡可识别两个单倍型之间的遗传顺式调控差异,但ASE数据可能存在偏差,可通过将RNA-seq读段比对到个性化参考基因组或聚合多个aseSNP的信号来减轻。ASE数据可用于提高识别eQTL的统计功效、定位因果调控变异,还可用于研究基因 - 环境相互作用和罕见遗传变异对基因表达的影响,提高孟德尔疾病的诊断准确性。
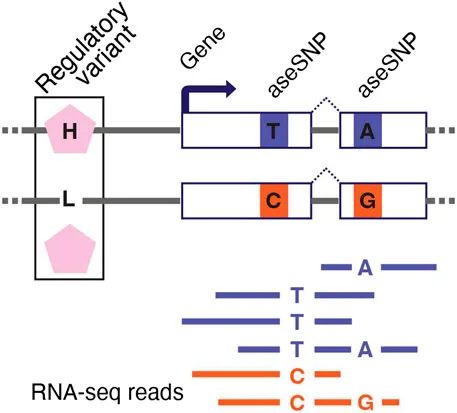
图3 用RNA-seq测量等位基因特异性表达:RNA-seq可用于生成具有转录区域杂合单核苷酸多态性(aseSNP)基因的等位基因特异性表达(ASE)数据。aseSNP使测序读段能够映射到其来源的单倍型。ASE数据中的不平衡是由杂合调控变异驱动的两个单倍型之间顺式调控差异的功能指标。多个aseSNP的数据可以聚合以提高ASE数据质量。这里所示的非编码调控变异有两个等位基因,分别诱导目标基因的高(H)和低(L)表达。
用RNA-seq分析环状RNA
环状RNA(circRNA)具有重要生物学功能,近年来相关计算工具不断涌现。circRNA的识别基于检测跨越环化接头(即反向剪接接头,BSJ)的读段,多数工具借助比对器检测潜在的反向剪接事件,部分剪接感知比对器可直接比对环状读段并检测BSJ。由于多数circRNA源于外显子区域,难以区分线性和环状读段,因此BSJ读段计数是衡量circRNA表达水平的可靠指标。此外,还有基于伪比对的工具可提高计算效率,通过计算接头比率可比较circRNA与其宿主基因的表达水平。针对circRNA的分析,还开发了用于转录本组装、内部结构可视化、差异表达分析的工具,以及多个综合数据库用于注释和优先级分析。
环状RNA是一类具有共价闭合环状结构的RNA分子。常用分析工具包括:
• CIRI:基于split-alignment的circRNA鉴定工具
• Find_circ:基于junction reads的circRNA检测工具
• CircAtlas:大规模circRNA数据库
讨论
随着技术的进步,RNA-seq 方法变得越来越流行,并且通过生物信息学社区不断努力开发准确且可扩展的计算工具,彻底改变了现代生物学和临床应用。此外,测序技术的进步提供了前所未有的能力来分析广泛的生物数据,使研究人员能够探索新的和现有的生物学问题。为了增加新用户和年轻科学家对 RNA-seq 方法的访问,本文概述了 RNA-seq 及其相关计算方法的基础知识,并讨论了各种应用的优缺点。RNA-seq 数据的计算分析可用于解决重要的生物学问题,例如估计各种表型和条件下的基因表达谱或检测特定外显子上的新替代剪接。RNA-seq 数据的专门分析还可以帮助检测影响剪接的转录因子的浓度、功能或定位的变化,可能导致神经退行性疾病和癌症的发生。一些最近开发的计算工具甚至能够重新利用 RNA-seq 数据来表征个体适应性免疫组和微生物组。此外,计算去卷积可以应用于 RNA-seq 数据以研究组织样本中的细胞类型组成。RNA-seq 应用和相关分析方法及软件开发的多学科性质引入了许多术语,这些术语可能会挑战更广泛的科学和医学研究社区的研究人员。RNA-seq 方法的文献传统上假设读者熟悉 RNA-seq 及其相关生物信息学分析的基本概念。这些方法可能需要多样化的计算技能才能有效使用。因此,缺乏计算技能可能会限制生物医学研究人员解锁 RNA-seq 的全部潜力,突显了解释基本 RNA-seq 概念并定义学科特定术语的综述的需求。
本文总结
RNA-seq数据分析是一个复杂但强大的过程,涉及多个步骤和工具。通过严格的质量控制、准确的读段比对、可靠的差异表达分析和深入的功能注释,研究人员可以从RNA-seq数据中提取有价值的生物学信息。
推荐阅读
中国银河生信云平台(网址:UseGalaxy.cn)以“让生信分析更简单”为使命。平台致力于为科研工作者、医疗机构和生物产业技术人员提供全栈式生物信息学分析解决方案。
优先技术响应、定制化工具部署、阶梯式能力培养,请加入「Galaxy生信星球」。咨询微信:usegalaxy 或 galaxy-help


















 2447
2447

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









