






点击蓝字 关注我们
分离培养的毛螺菌科潜在新种拓展了基因组和功能多样性

iMeta主页:http://www.imeta.science
研究论文
● 原文链接DOI: https://doi.org/10.1002/imt2.174
● 2024年2月13日,深圳华大生命科学研究院邹远强、肖亮团队在iMeta在线发表了题为 “Isolation of potentially novel species expands the genomic and functional diversity of Lachnospiraceae” 的文章。
● 本研究揭示了毛螺菌科新物种尚未发掘的潜力,基于此选择菌株,得以开发有望改善人类健康和疾病管理的下一代益生菌。
● 第一作者:林晓倩
● 通讯作者:Karsten Kristiansen(kk@bio.ku.dk),肖亮(xiaoliang@genomics.cn),邹远强(zouyuanqiang@genomics.cn)
● 合作作者:胡童远,吴智楠,李灵讷,王钰淏,文定洋,刘旭东,李文曦,梁何伟,金鑫,徐讯,汪建,杨焕明
● 主要单位:深圳华大生命科学研究院
亮 点

● 全面的基因组分析揭示了毛螺菌科被低估的物种多样性和独特功能;
● 1.5 M基因集合的构建突显了毛螺菌科在人类肠道微生物群中的重要作用,以及潜在新物种的贡献;
● 毛螺菌科在短链脂肪酸(SCFA)合成、产次生代谢产物和形成孢子方面表现出巨大的潜力,具有广阔的研究和应用前景;
● 毛螺菌科的丰度与动脉粥样硬化性心血管疾病(ACVD)和炎症性肠病(IBD)等疾病相关,表明其具有潜在的保护作用。
摘 要
毛螺菌科有望成为下一代益生菌,但目前缺乏对其多样性的全面描述,阻碍了可应用的菌株的选择。为了解决这一不足,我们将公共数据库与我们的新分离株相结合,对1868个高质量基因组进行了深入的基因组和功能分析。该数据集代表387个定植选择性的物种级簇,其中8个属为多谱系。泛基因组分析、单核苷酸多态性(SNP)鉴定和益生菌功能预测表明,物种分类、栖息地和地理位置共同塑造了毛螺菌科的功能多样性。此外,对动脉粥样硬化性心血管疾病(ACVD)和炎症性肠病(IBD)的关联分析表明,几种潜在的新型毛螺菌科物种具有减少机会性病原体丰度的潜力,从而带来潜在的健康益处。我们的研究结果揭示了新物种尚未发掘的潜力,基于此选择菌株,得以开发有望改善人类健康和疾病管理的下一代益生菌。
视频解读
Bilibili:https://www.bilibili.com/video/BV1oz421Q7rT/?vd_source=bd1effa8c29673890a66968682d86f6c
Youtube:https://youtu.be/x0c2xgrre_I
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
毛螺菌科(Lachnospiraceae)是芽孢杆菌门的一个科,由几个严格厌氧的属组成,它们在哺乳动物(特别是人类和反刍动物)的肠道中含量丰富,此外该科的成员也在环境中定植。先前的研究根据宏基因组数据揭示了毛螺菌科成员与多种疾病之间存在显著关联,此外,在患有动脉粥样硬化性心血管疾病(ACVD)的个体中发现Roseburia spp.显著减少。毛螺菌科中的许多物种都具有重要的功能,例如胆汁酸转化、短链脂肪酸合成和人类胃肠道中的抗生素生产,并且许多成员被报道对人类健康具有有益影响。先前研究发现,口服Anaerobutyricum soehngenii可以改善代谢综合征患者的胰岛素敏感性,Anaerobutyricum hallii可以改善2型糖尿病患者的餐后血糖控制。因此,毛螺菌科似乎有望成为下一代益生菌的候选。
然而,研究还表明毛螺菌科的某些菌株可能会促进疾病的发展。例如,从口腔中分离出的Catonella morbi ATCC 51271被认为与牙周炎有关,Anaerostipes hadrus BPB5已被证明会加重葡聚糖硫酸钠诱导的小鼠的结肠炎,从人类血液中分离出的 Eisenbergiella tayi甚至可以作为机会性病原体。因此,选择合适的物种或菌株对于未来的临床前研究极为重要。先前对毛螺菌科 5个属的分离株进行分析的研究表明,人类来源的分离株之间存在高度多样性,但在《原核生物命名标准列表》(LPSN,https://lpsn.dsmz.de/,截至2021年7月)中,毛螺菌科至少包含58个属和122个有效且正确命名的物种。因此,尽管大量研究已经验证了毛螺菌科对宿主健康的影响,但大多数感兴趣的分类单元仍缺乏物种水平的分类,这意味着存在大量潜在的新物种,需要对毛螺菌科的基因组多样性进行进一步研究。
在我们之前扩大的培养细菌参考基因组集合(称为CGR2)的研究中,我们从中国健康成年人的粪便中培养了756个毛螺菌科菌株,并生成了高质量的基因组。通过从公共数据库收集可用的基于培养的基因组数据,并将这些数据与CGR2的基因组相结合,我们构建了一个包含1868个毛螺菌科的高质量基因组的集合。这些基因组揭示了毛螺菌科显著增加的分类多样性,并且潜在的新物种极大地扩展了现有的基因和功能概况。此外,全面的毛螺菌科培养基因组集合提高了疾病相关标志物的分辨率,并为选择对人类健康具有潜在有益影响的菌株提供了基础。
结 果
CGR2增加了毛螺菌科的分类学多样性
我们从CGR2中获得了756个高质量的毛螺菌科基因组,为了评估这些基因组的新颖性,我们从LPSN检索了58个属和122个有效命名的物种,并下载了它们的16S核糖体RNA(rRNA)基因序列作为参考。分别使用98.7%和94.5%的相似度作为种和属的划分,我们发现CGR2中新分离培养的基因组中有47.88%是潜在新种,22.22%是潜在新属。此外,潜在新属的16S rRNA基因序列被聚类为37个属级别操作分类单元(OUT)和64个物种级别OTU。值得注意的是,CGR2的基因组不仅涵盖了人类肠道微生物群中发现的毛螺菌科的大多数属,而且还添加了三个以前从未从人类肠道中分离出来的潜在新属(图 1A)。总之,这些结果表明了毛螺菌科的分类多样性,但仍需要进一步研究来充分探索毛螺菌科的多样性。
接下来,我们从NCBI、IMG和UHGG(截至2021年8月)收集了毛螺菌科的分离培养基因组,结果CheckM质量控制产生了1868个高质量基因组,其中包括来自CGR2的756个基因组。这些基因组来自六大洲(非洲、亚洲、欧洲、北美洲、大洋洲和南美洲)总共32个国家的人类、动物和环境等多个来源的分离菌株,这凸显了毛螺菌科在不同地区和国家的普遍分布(图1B和表S1)。这一结果与早期宏基因组数据中毛螺菌科的流行率和丰度一致,表明毛螺菌科的成员在哺乳动物胃肠道和环境样本中很常见。
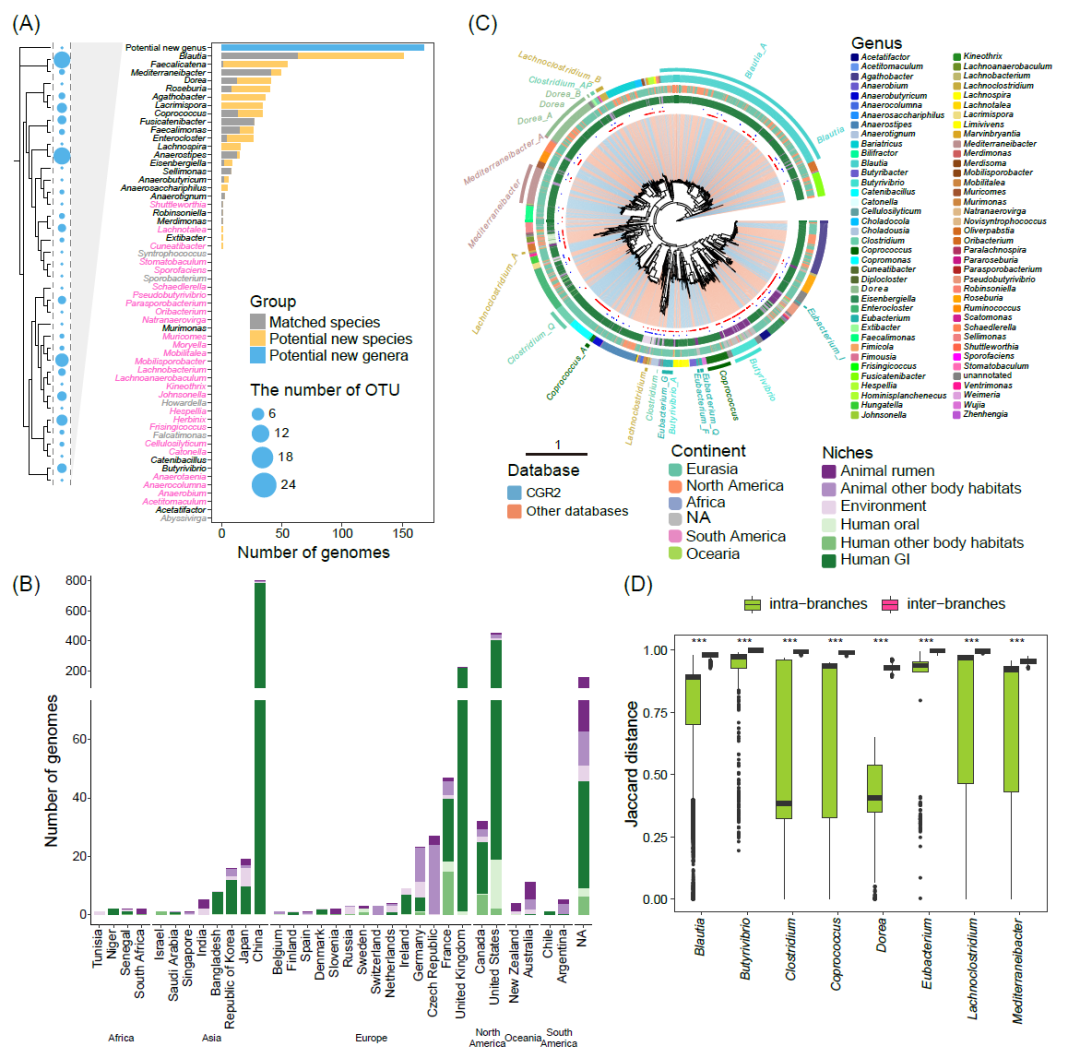
图 1. 1868个毛螺菌科培养基因组的集合
(A)756个新分离的毛螺菌科基因组对现有有效命名属的贡献。有人类肠道培养基因组代表的属标记为黑色,没有人类肠道培养代表但具有来自其他生态位的基因组的属标记为粉色,没有任何培养基因组的属标记为灰色。(B)收集的基因组在不同地理和生态位分布情况。(C)1868个分离基因组的系统发育树。该树是使用 PhyloPhlAn 3从串联蛋白质序列生成的。进化枝根据基因组源数据库(CGR2或下载)进行着色。潜在的新物种和模式菌株在第一层中分别用红点和蓝点标记。第二层和第三层分别代表基因组初始分离的生态位和大陆。GTDB属注释标记在最后一层,并为具有多个谱系的物种进行文本标记。(D)分支内和分支之间的八个多谱系属的Jaccard距离分布。***p < 0.001,由Wilcoxon检验定义。
系统发育分析揭示了系统发育多样性和定植选择性
基于95%平均核苷酸同一性(ANI)阈值,1868个基因组被聚类为387个物种级簇,超过之前注释的物种数量的三倍(表S1和S2)。数字DNA-DNA杂交(dDDH)是另一种用于估计两个细菌基因组之间的遗传相关性或相似性的生物信息学技术,簇内和簇间的dDDH值支持我们研究中当前对物种级簇的描述(图S1)。许多属,包括Blautia、Copromonas、Butyrivibrio、Coprococcus和Pseudobutyrivibrio,都含有大量潜在的新物种(图1C和表S1)。在Blautia、Butyrivibrio、Clostridium、Coprococcus、Dorea、Eubacterium、Lachnoclostridium和Mediterraneibacter这8个属中,我们观察到系统发育树中至少有两个明显的分支(图1C),遗传组成证实了这些分支之间的分歧(图1D)。值得注意的是,分支间的Jaccard值相当高且分布更窄,中值范围为93.03%至99.86%(图1D),这些值显着高于分支内观察到的值,这表明分支间存在更大的遗传变异性,支持它们适当分组的潜在理由。
ANI和16S rRNA基因序列相似性是原核生物物种划分的两种常用策略,但这两种方法可能会产生偏差。因此,我们研究了两种方法之间物种划分的差异。以109个已测序的模式菌株基因组和16S rRNA基因序列为参考,我们发现了26个物种级簇,其16S rRNA基因序列保守,但基因组高度多样化,被GTDB注释为新物种或多系谱系(图S2)。相反,7个物种水平簇与其模式菌株表现出较低的16S rRNA基因相似性(图S2),我们将模式菌株基因组预测的16S rRNA基因序列与下载的序列进行了比对,得到了相同的结果。因此,有必要对这7个物种级簇的16S rRNA基因序列可能存在的污染进行彻底分析。
我们收集的基因组代表了从人类、动物和环境中分离出来的物种,证实了之前的研究结果。基于培养方法,我们可以在物种甚至菌株水平上追溯起源。我们发现75个属中有30个属是从不同的生态位中分离出来的,主要是人类和动物,而93.02%的物种级簇是从特定的生态位中分离出来的,这表明毛螺菌科特定成员的选择性定植(图S3)。
毛螺菌科基因组中的基因和蛋白质组
为了建立功能图谱,我们构建了基于1868个基因组的基因目录。结果表明,来自CGR2的培养物种的新基因组将毛螺菌科基因目录扩展至1.5M(图2A)。我们研究了潜在新物种对基因目录的贡献,发现这些迄今为止未知的物种贡献了42.34%的基因 此外,编码甲基接受趋化蛋白、内切葡聚糖酶、肽/镍转运系统底物结合蛋白和鞭毛蛋白的基因在潜在的新物种中富集,表明这些物种可能为葡萄糖代谢和运动性等方面提供新的见解(图S4B)。
对于这些基因编码的蛋白质序列,我们构建了蛋白质序列目录。与基因目录类似,我们获得了1.4M的蛋白质目录(图S4A),而其中超过半数(55.52%)是Prokka数据库注释的假设蛋白质。
我们试图探究毛螺菌科成员对人类肠道微生物群功能的影响,这使得我们提取了分离培养自人类胃肠道的毛螺菌科基因组中预测的蛋白质序列。随后,我们使用统一人类胃肠蛋白目录(UHGP)分析了这些序列,该目录被誉为人类肠道微生物组中最全面的蛋白质存储库。结果显示,从人体胃肠道中分离出来的毛螺菌科成员覆盖了人体肠道菌群近50%的功能,包括代谢、遗传信息处理和环境信息处理(图2B)。
鉴于肠道微生物在宿主营养和代谢中发挥的关键作用,我们的分析重点是源自人类肠道的毛螺菌科成员的代谢功能。我们表明这些成员不仅具有强大的碳水化合物代谢、脂肪酸合成和降解能力,而且还参与支链氨基酸生物合成、嘌呤和尿素代谢以及叶酸生物合成,这些对于宿主生理调节具有重要意义(图2C和图S4C)。此外,人类肠道毛螺菌科的成员贡献了59个独特的KEGG Orthologies(KO)(图S4D),主要涉及合成和代谢功能。
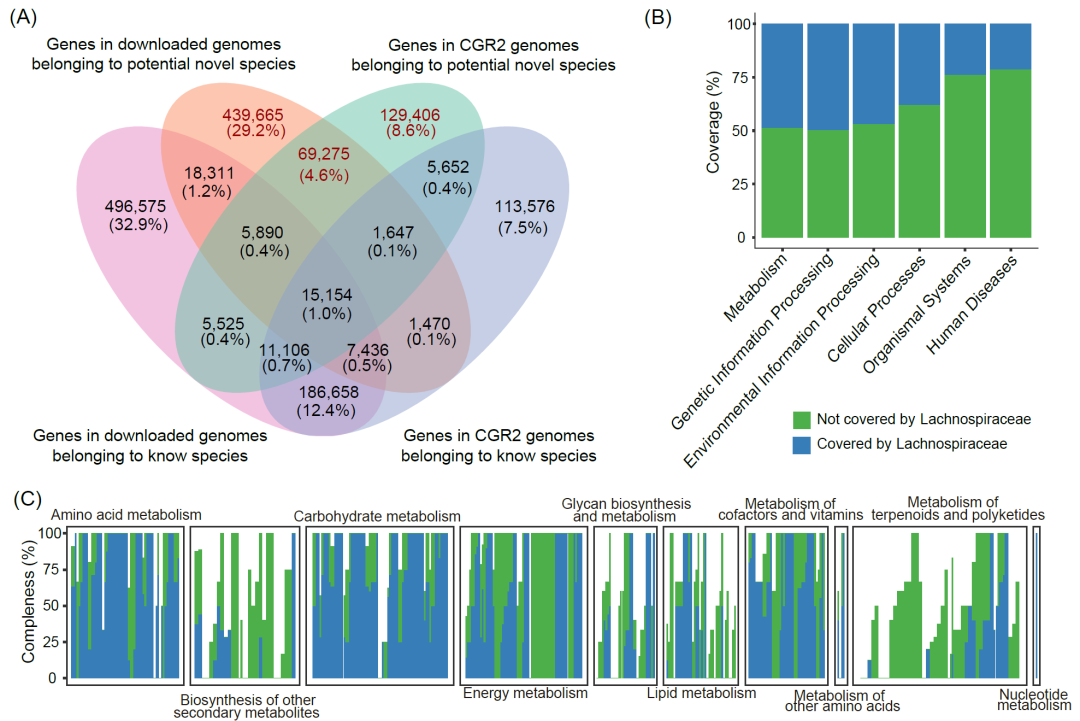
图 2. 对毛螺菌科基因的新见解
(A)1.5 M毛螺菌科分离株的基因目录,潜在新物种基因组特有的部分以红色突出显示。(B)人类肠道毛螺菌科基因在人类肠道微生物群功能的百分比。(C)各代谢功能模块的完整性。一个条代表一个模块,所有模块根据功能可以分为10类。
泛基因组分析揭示了毛螺菌科代表的生态多样性
泛基因组代表了进化枝内所有物种或菌株的整套基因,从而表征了基因组之间的多样性,并为进化起源和生态位适应提供了重要见解。分离株的全基因组测序为鉴定密切相关菌株之间的核心和独特基因奠定了基础。
首先,通过使用毛螺菌科的所有基因组构建科级别泛基因组,我们发现最普遍的基因仅由41.54%的基因组共享,而几乎99.99%的基因(被定义为云基因)仅分布在少数几个基因组中(图3A)。我们接着分析了包括至少10个独立的基因组的属和种的泛基因组和核心基因组大小。在属水平上,核心基因组和泛基因组的大小与基因组和簇的数量呈正相关。泛基因组的范围变化高达10倍,而核心基因组的范围变化超过1000倍(图S5A)。在物种水平上,Hungatella effluvii、Eisenbergiella tayi、Enterocloster boltteae和Enterocloster clostridioformis拥有更多的基因,其核心和泛基因组规模也更大。此外,基因组数量最多的前10个物种具有较小的核心基因组和较大的泛基因组,表明基因组的遗传组成更加多样化(图3B)。
Blautia wexierae拥有较大的泛基因组和相对较小的核心基因组,然而,它缺乏单核苷酸多态性(SNP)分析所需的完整参考基因组。因此,我们选择了Agathobacter rectalis,它拥有第二大的泛基因组和完整的参考基因组。我们进一步以分离自法国的完整基因组GCA_000020605为参考,对分离自5个国家的96个基因组进行了SNP分析。我们在基因内区域检测到95173个突变,其中19357 个是错义突变。SNP的系统发育树显示了与地理位置相关的四个进化枝(图3C)。最接近参考基因组的是Clade 1,由分离自美国的基因组组成;其次是Clade 2,主要由分离自英国的基因组组成;Clade 3由分离自多个国家的基因组组成;Clade 4主要由分离自中国的基因组组成(图3C)。在注释了位于基因内区域的变体后,我们重点关注基因bceB,它编码参与杆菌肽输出的ABC转运蛋白复合物BceAB,并包含大量错义突变和一个影响很大的突变(图S6A)。四个分支表现出不同的bceB基因突变模式。Clade 4中与bceB相关的突变最高,而Clade 2的突变最低(图S6B)。Clade 1中的频繁突变位于基因的后端,而Clade 3中的频繁突变则位于基因的前端。此外,所有四个分支都有一种导致翻译提前终止的突变,尤其是Clade 3,在82.14%的基因组中发现了突变(图3D)。ABC转运蛋白BceAB 介导对羊毛硫肽、杆菌肽和β-内酰胺抗生素等抗菌肽的耐药性。该结果表明Agathobacter rectalis表现出不同程度的抗性丧失。
为了研究潜在新物种对属内多样性的影响,我们重建了五个属的泛基因组累积曲线(图S5B)。大量潜在新物种的添加提供了更大的泛基因组。与已知物种相比,新物种具有更广泛的功能潜力。例如,与之前已知的物种相比,Butyrivibrio的泛基因组增加了两倍多(图S5C)。
对Anaerotignum(同时存在于人类、动物和环境中的一个属)的核心和特有基因的研究表明,不同的生态位已经进化为含有独特和生态位相关基因的不同物种(图3E)。
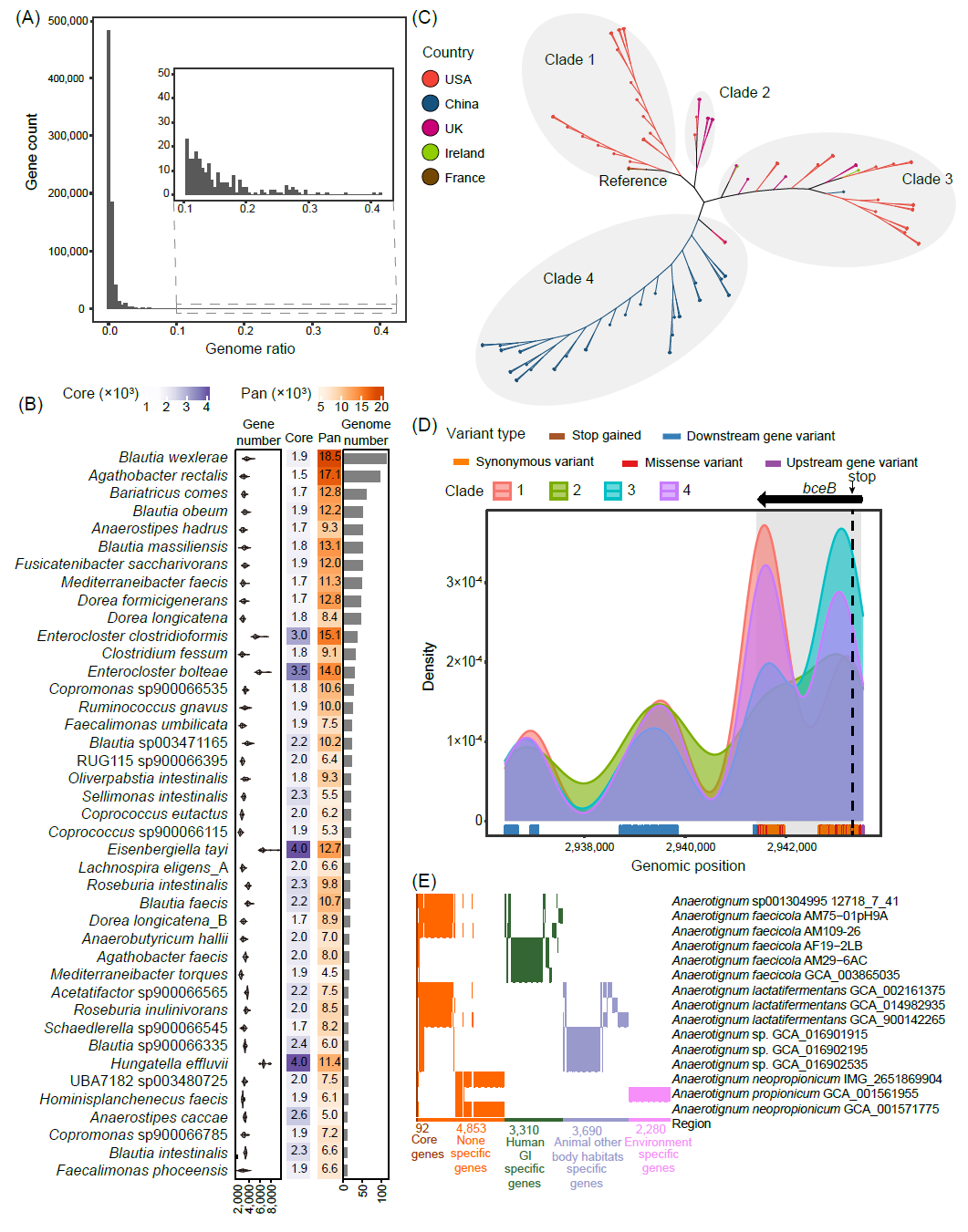
图 3. 毛螺菌科的泛基因组多样性
(A)不同基因组之间共享的基因数量。X轴是所有基因组中含有共同基因的基因组的比例。(B)10个以上基因组的41个物种的核心基因组、泛基因组、基因组数和基因数统计,按基因组数排序。(C)基于SNP构建的系统发育无根树,由Parsnp构建。节点根据分离培养国家/地区进行着色。根据遗传距离划分进化枝。(D)四个进化枝不同基因组位置的突变频率密度。基因组位置根据突变类型着色。(E)Anaerotignum属中基因的有无情况。仅存在于特定生态位的基因组中的基因被定义为特定基因。
物种分类、身体栖息地和地理的多样性塑造了毛螺菌科的各种功能
人类肠道微生物群将碳水化合物发酵成短链脂肪酸(SCFA),尤其是丁酸和丙酸,然后被宿主利用。SCFA为肠上皮细胞提供能量,调节免疫系统,并影响维持宿主健康所必需的各种代谢途径。毛螺菌科的成员被认为是肠道SCFA的主要生产者。丁酰辅酶A产生丁酸有两种不同的途径,一种依赖于丁酸激酶,另一种依赖于丁酰辅酶A:乙酸辅酶A转移酶(表S3)。丙酰辅酶A转化为丙酸包括三种不同的途径,包括由辅酶A转移酶或辅酶A连接酶催化的一步反应和涉及多个中间步骤的途径(表S3)。我们发现,只有40.80%的 毛螺菌科基因组具有完整的丁酸途径,而几乎所有基因组都具有需要 CoA转移酶的完整丙酸途径(图4A)。此外,毛螺菌科基因组中预测的完整丁酸途径通常取决于丁酸激酶或丁酰辅酶A:乙酸辅酶A转移酶,而毛螺菌科的同一基因组上携带一个或多个完整的丙酸途径。Coprococcus是一种公认的丁酸产生菌,不仅具有完整的丁酸途径,而且还具有不同的丙酸途径,显示出其生产 SCFA的巨大潜力。我们还发现大量潜在的新物种、新属具有生产丁酸和丙酸的能力。
将包含完整丁酸途径的成员的属定义为潜在的丁酸生产属,我们从26个属中探究了与丁酸生产相关的基因的分布(图4B)。我们发现该通路的完整性与生态位无关,而是取决于物种。同一属内的不同谱系在途径完整性和途径类型上表现出差异,即在属水平上不存在特异性。例如,Coprococcus catus使用丁酰辅酶A:乙酸辅酶A转移酶来产生丁酸,而Coprococcus的其他基因组则使用丁酸激酶途径。对于Enterocloster,E. clostridioformis的一些基因组使用丁酰辅酶A:乙酸辅酶A转移酶,而Enterocloster bolteae的个别基因组含有编码可通过两种途径产生丁酸的酶的基因,但其余基因组具有基因缺失。这一结果与系统发育不相符,与丁酸生产相关的基因可能是通过水平基因转移获得的。
许多研究表明毛螺菌科成员可产生新型次级代谢产物。我们使用antiSMASH(V6.0.0)对次级代谢产物生物合成基因簇(SMBG)进行了广泛的挖掘,并从1856个基因组中探索了6688个基因簇(7373个SMBG),总共32种类型(表S4)。通过将这些基因簇与MiBIG数据库中经过实验验证的参考生物合成基因簇(BGC)进行匹配,我们在毛螺菌科基因组中鉴定出了58个已知BGC(图 4C)。数量最多的BGC是二肽醛、瘤胃球菌素A(RumA)和胞外多糖。二肽醛是一种高效的蛋白酶抑制剂,最初在Ruminococcus sp.中发现。RumA可用于临床治疗致病性Clostridium spp.感染,之前已在E. gnavus E1中进行了表征。我们发现Blautia具有生产二肽醛和RumA的巨大潜力,这不仅有助于它们占据生态位,而且可以作为这些生物活性产品的重要候选来源。胞外多糖主要由Anaerostipes产生,可作为交叉喂养地底物刺激特定有益菌的生长,减少病原体粘附,提高肠道屏障的保护作用。此外,90.7%的基因簇在MiBIG数据库中没有匹配,表明其结构和功能尚未被描述。这些结果表明,毛螺菌科在发现新型次级代谢产物方面具有巨大的未开发潜力。
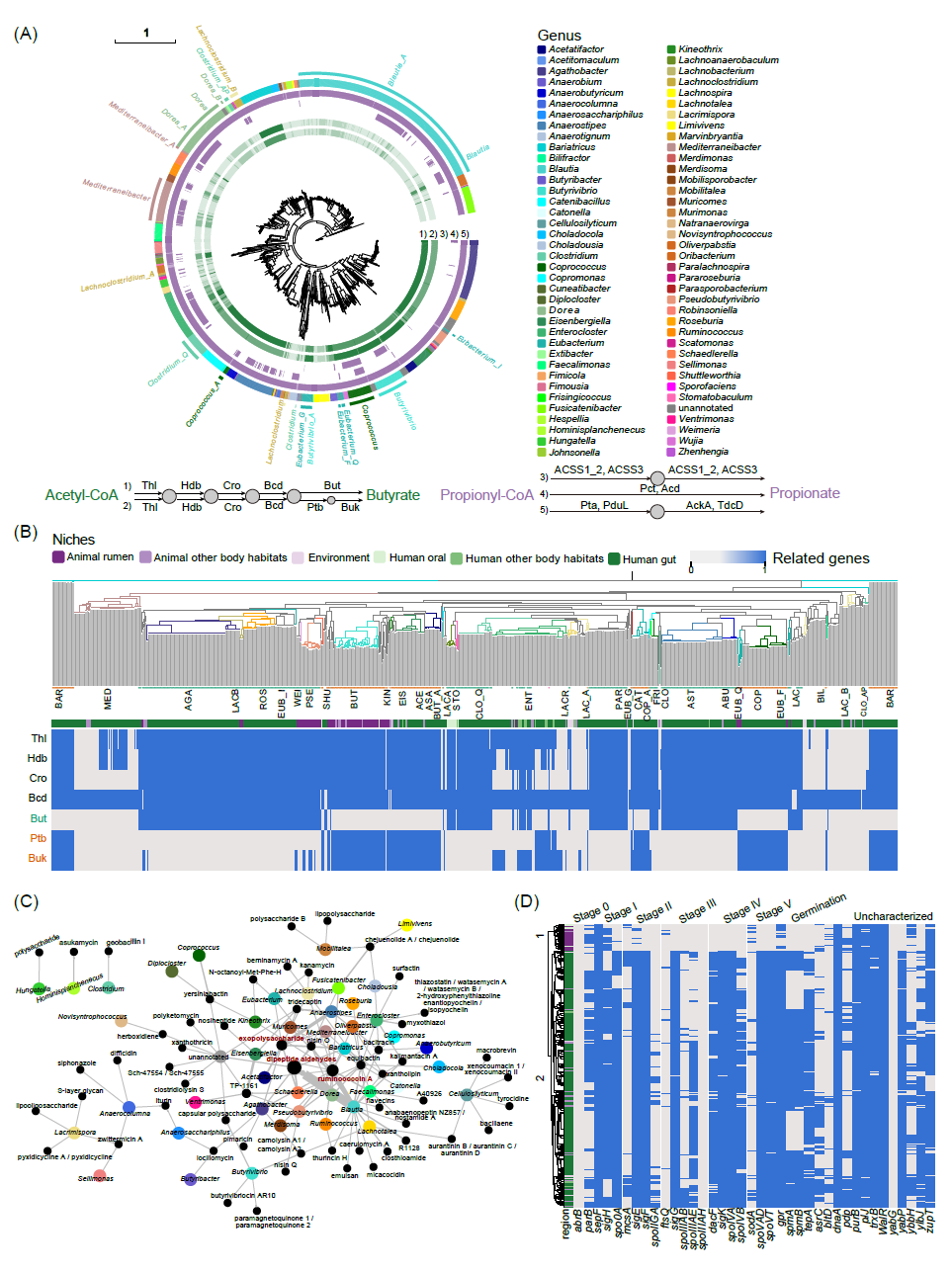
图 4. Lachnospiraceae 的功能概况
(A)每个基因组中与短链脂肪酸产生相关的基因的分布。该系统发育树与图1C一致。乙酰辅酶A产生丁酸的两条途径(丁酰辅酶A转移酶途径和丁酸激酶途径)分别出现在第一层和第二层。第三层到第五层代表从丙酰辅酶A到丙酸盐生产的三个途径。颜色的深浅表示通路中基因的类型,即通路的完整性。最后一层代表属,与图1C一致。(B)具有产生丁酸潜力的属的系统发育树。进化枝的颜色代表属,第一层代表基因组的生态位。热图根据基因的存在或不存在进行着色,对应于与左侧显示的两条通路相关的基因。(C)属与已知次级代谢物之间的关系网络。属用相应颜色的点表示,次级代谢物用黑点表示,点的大小与数量有关。(D)孢子形成基因分布的热图。
孢子介导的传播在不同的生态位之间存在差异
大多数Bacillota是已知的产孢子细菌,它们在恶劣的环境条件下(例如高温或低温、寡营养条件和接触药物)表现出长期活性。此外,孢子还可以促进细菌在宿主之间传播,最终在多种生态位定植。大多数毛螺菌科物种被认为是孢子生产者,毛螺菌科的孢子已被证明能够在乙醇处理下存活并在胆汁酸存在的情况下发芽。Browne等人使用机器学习方法识别了66个孢子形成特征基因,并进一步显示了这些基因在不同谱系的肠道Bacillota中的差异丧失。我们发现分离培养自动物瘤胃和人类口腔的基因组中存在大量特征基因的缺失(图4D,图S7)。然而,尽管来自人类胃肠道的基因组包含相对完整和多样的特征基因,但其中一些基因组丢失了孢子形成所必需的关键调节基因spo0A。这种差异可能表明不同生态位之间的适应。
毛螺菌科与人类疾病的关联
基于宏基因组学的方法可以识别疾病相关标志物,但主要依赖于基于数据库的reads注释或de novo分箱,通常缺乏物种水平的匹配,从而限制了后续研究。此外,由于大多数生物标志物缺乏培养菌株,宏基因组研究识别的与疾病相关的细菌种类其功能信息和机制见解有限。为了规避这一限制,我们使用培养基因组来识别健康与疾病之间的潜在关联。
使用我们的基因组集合对中国ACVD队列进行分析,我们揭示了细菌菌株丰度的显著差异。具体来说,我们从13个物种级簇中识别出56株菌株,它们在健康对照组中明显更为普遍,而来自14个簇的52株菌株在ACVD组中表现出显著富集(调整后的p值 < 0.01且|log2 FC| > 1,图S8A表S5)。与之前的研究一致的,我们观察到健康对照组中Roseburia属成员的丰度较高,而被认为是机会性病原体的Ruminococcus gnavus在ACVD组中显著富集。此外,我们发现Lachnospira eligens、Acetatifactor sp.和Agathobacter faecis的几种菌株不仅在共丰度网络中发挥着关键作用,而且还与ACVD富集的基因组表现出负相关性(|r| > 0.3,调整后的p < 0.01,图S8B)。
对于肠道炎症性疾病,我们探究了毛螺菌科成员在人类微生物组计划(HMP)炎症性肠病(IBD)队列中的分布,包括克罗恩病(CD)和溃疡性结肠炎(UC)患者。在CD患者中有8个簇的17株菌株富集,而来自35个簇的122株菌株丰度减少(调整后的p值 < 0.01且|log2 FC| > 1,图S9A和表S6)。在健康组中,我们发现了潜在的新物种的富集,例如Acetatifactor sp.、Choladocola sp.、Eubacteria sp.和来自未知属的菌株。这些新物种与Enterocloster bolteae和R. gnavus表现出显著的负相关性(|r | > 0.3,调整后的p < 0.01,图S9B)。在UC患者中,富集了来自7个簇的8株菌株,而减少了来自24个簇的51株菌株(调整后的p值 < 0.01且|log2 FC| > 1,图S10和表S7)。有趣的是,两株菌株,GCA_009881395和GCA_013304625,它们都是Blautia wexlerae,但显示出不同的分布模式:GCA_009881395在健康对照组中富集,而GCA_013304625在UC组中富集(图S11)。这一观察结果表明,来自同一物种的不同菌株可能在健康和疾病中具有不同的作用,强调了在宏基因组疾病研究中考虑菌株水平多样性的重要性。
比较这两个队列,我们观察到健康组中富集的菌株数量存在差异。虽然ACVD队列显示出更高丰度的特定菌株,但IBD队列却显示出更多数量的富集菌株(图S11)。这些对比发现强调了识别与不同疾病相关的独特微生物特征的重要性,并强化了在疾病宏基因组研究中进行菌株水平分析的重要性。
方 法
CGR2基因组新颖性评估
我们在本研究开始时,即2021年7月,下载了LPSN中所有有效且正确命名的122个模式菌株的16S rRNA基因序列。使用Barrnap(v0.9)提取了756个CGR2基因组的16S rRNA基因序列。BLAST 2.12.0+用于进行配对BLASTn,物种水平的同一性阈值为98.7%,属水平的同一性阈值为94.5%。CGR2中潜在新属的16S rRNA基因序列通过usearch(v11.0.667)进行聚类,以获得种和属水平的OTU(分别使用参数:‐‐id 0.987和0.945)。MAFFT(v7.310)用于提取和比对属级代表性16S rRNA基因序列,trimAl(v1.4 rev 22)进行修剪。使用FastTree(v2.1.3 SSE3)的最大似然法重建系统发育树。
基因组收集和质量评估
为了建立毛螺菌科的分离基因组集合,我们从NCBI和IMG数据库(2021年7月)下载了标记为毛螺菌科的所有分离培养基因组,并且纳入当前建立的CGR2和UHGG已培养基因组集合。接着对所有基因组进行了信息收集,包括宿主(动物栖息地包括牛、羊、小鼠、猪、鸡、狗、羊驼、小袋鼠和木雕水龟)和国家信息。为了避免数据库同步导致的基因组重复,我们使用fastANI(v1.32)来比较不同数据集的基因组,当基因组高度相似时(pairwise ANI为100%)且菌株名称相同时只保留其中一个基因组。只有由CheckM(v1.1.2)估计的完整性>90%且污染<5%的基因组才被定义为高质量基因组,并保留用于进一步分析。
系统发育和分类学分析
大于等于95% ANI的基因组被认为是同一物种。因此,我们使用fastANI(v1.32)来计算基因组之间的成对ANI值并生成矩阵(‐‐matrix)。随后,我们使用R中的“hclust”函数(method=“complete”)进行了层次聚类分析,然后使用“cutree”函数根据95% ANI阈值(h=0.05)将所得的层次聚类树状图划分为簇。dDDH值由GGDC计算(https://ggdc.dsmz.de/ggdc.php#)。使用GTDB-Tk(v2.3.2,数据库版本214)的“classify_wf”函数和默认参数对每个基因组进行分类注释,任何没有有效名称的谱系都被认为代表一个潜在的新物种或属,属名的附加字母后缀表明高度的系统发育多样性。PhyloPhlAn 3.0用于对1868个基因组进行系统发育分析,该过程涉及几个具体步骤。最初,DIAMOND被用来识别标记基因,随后通过MAFFT处理映射结果以进行对齐优化,trimAl用于进一步细化比对后,通过IQ-TREE构建最大似然树,最后使用RAxML进行细化。本研究中的所有系统发育树均使用在线工具EVOLVIEW v2进行可视化和注释。
非冗余基因/蛋白质目录的构建和泛基因组分析
在我们分析的初始阶段,1868个基因组由Prokka v1.14.6使用默认设置进行注释,以预测与这些基因组中存在的基因相关的核苷酸和蛋白质序列。CD-HIT v4.6.3使用核苷酸序列来生成基因目录,特定参数(‐c 0.95和‐aS 0.9)定义了95%蛋白质同一性和90%覆盖率的严格标准。此步骤有助于生成非冗余基因目录,确保每个基因都有唯一的表示并消除数据集中的冗余。与此同时,我们还进行蛋白质目录的构建,蛋白质序列作为MMseq2(版本13.45111)的“linclust”功能的输入,采用一组定义的参数,包括“--cov‐mode 1 ‐c 0.8 --kmer‐per‐seq 80 ‐‐min-seq‐id 0.95”,定义了95%蛋白质同一性和80%覆盖率的标准。泛基因组分析由Roary v3.7.0进行,参数“‐i 90”来识别科、属和物种的核心基因和云基因。对于8个多谱系属,提取了Roary生成的基因-基因组矩阵,其中包括所有基因组中每个基因家族的存在/不存在概况,并使用R函数“vegdist”计算基因组之间的成对Jaccard指数。热图可视化由R包ComplexHeatmap进行。采用默认设置的Parsnp v1.5.0从Agathobacter rectalis的所有基因组中分析SNP,并生成基于SNP的系统发育树。使用SnpEff v5.1对突变进行注释,报告了它们对蛋白质的预测影响(HIGH、MODERATE、LOW或者MODIFIER)。
功能表征
非冗余基因目录的功能概况由eggNOG-mapper v2(eggNOG数据库版本:5.0.2)进行。KEGG ORTHOLOGY(KO)是从eggNOG-mapper结果中提取的,并在iPath3中可视化。为了识别已知簇和新簇之间存在显着差异的KO,使用Huttenhower Lab Galaxy模块确定了线性判别分析效应大小(LEfSes),每个基因组的基因值总和被标准化为1M后,输入参数如下:子类间成对Wilcoxon检验的α值为0.01,判别特征的对数线性判别分析得分阈值为2.0。根据之前的研究生成乙酰辅酶A至丁酸、丙酰辅酶A至丙酸的生物合成途径,并从KEGG数据库中提取相关酶的蛋白质序列以构建小型数据库。相关酶的酶委员会编号显示在表S3中。为了更好地描述菌株参与丁酸和丙酸产生的潜在能力,我们对上面构建的数据库中每个菌株的基因序列进行了BLAST(blastp,cut-off 1e-2,同一性≥60%,覆盖度≥50%)。如果一个基因组可以注释上图3A中路径中所有步骤的酶,则它被定义为具有完整的路径。使用antiSMASH 6.0总共挖掘了7373个SMBG。使用Cytoscape(v3.8.2)显示已知功能SMBG与其来源基因组之间的关系。先前的研究提出了66个孢子形成特征基因,我们在prokka注释结果中搜索这些基因名称,以获得特征基因在每个基因组中的分布。
毛螺菌科疾病相关标记物的鉴定
ACVD队列的385个宏基因组的clean reads(171个来自健康个体,214个来自ACVD患者)从欧洲生物信息学研究所(EBI)数据库下载,登录号为 ERP023788。此外还下载了IBDMDB研究的人类肠道宏基因组测序数据(https://portal.hmpdacc.org/),metadata可以通过https://ibdmdb.org/results/HMP2/找到。为了计算样本中毛螺菌科基因组的丰度,我们建立了一个Kraken2/Bracken 数据库(选项:‐k 31 ‐l 100),其中包含毛螺菌科家族的1868个基因组。对于每个样本,使用Kraken2 v2.1.2和Bracken v 2.6.1执行reads分配,指定0.001%相对丰度和至少10%出现率的阈值来定义样本中基因组的存在。R用于计算丰度,并且过滤掉没有任何读数映射的样本或基因组。EdgeR用于识别丰度显著不同的基因组,并选择调整后的p值< 0.01和|log2 FC| > 1的基因组。R函数“corr. test”进行细菌共现分析。
讨 论
在这项研究中,我们整合了毛螺菌科的全面基因组集合,用于分析分类学和功能多样性。毛螺菌科的成员在生命早期就开始定植,并随着年龄的增长而增加。CGR2分离培养的基因组贡献了大量潜在的新物种,为毛螺菌科未经探索的分类多样性提供了初步指示,强调了基于培养的研究在揭示毛螺菌科分类丰富性和多样性方面的重要性。结合公开的基因组,我们发现毛螺菌科细菌在各个生态位中普遍存在,而物种水平的定植表现出特异性,为使用毛螺菌科成员的提供干预研究了宝贵的见解。与目前已表征的122个物种相比,目前分离培养的菌株使物种数量增加了三倍,这表明迄今为止毛螺菌科的物种多样性被低估了。值得注意的是,毛螺菌科的八个属表现出更大的多样性,在系统发育树中显示出不同的分支,以及SCFA合成途径和疾病关联的变化。我们建议有必要对潜在新物种的分类学进行研究并提供有效的名称,并且有必要对多谱系属进行重新分类以完善分类法。在功能探索方面,我们构建了一个1.5 M基因目录和一个1.4 M蛋白质目录,其中潜在的新物种发挥了主要作用,为毛螺菌科代谢和生物学提供了新的线索。人类肠道来源的毛螺菌科菌株涵盖了人类肠道微生物群中近一半的功能,进一步强调了它们在这个生态系统中的重要作用。我们分别对属和物种进行了泛基因组分析,丰富了我们对研究充分的成员和研究不足的成员的理解。针对所有基因组构建了SCFA的生物合成途径,并预测了大量SMBG,强调了毛螺菌科在人类肠道中的潜在生态重要性。研究表明,Blautia和Roseburia等通常被认为是有益物种的物种是SCFA的主要生产者。
尽管微生物次级代谢物的研究主要集中于环境中存在的特定生物体,特别是Streptomyces、Aspergillus和Pseudomonas,但宿主相关的微生物也可能产生有趣的次生代谢物。在MiBIG数据库的1926个经过实验验证的次级代谢物中,只有10个是最初从毛螺菌科的四个属中发现并提取的。我们的研究预测来自1856个基因组的7373个基因簇有可能产生有趣的次生代谢物,这表明毛螺菌科具有产生此类化合物的惊人能力,其中大部分仍有待表征。
孢子是由Bacillota形成的抗逆结构。近年来随着人类肠道微生物培养组学的发展,越来越多的菌株和高质量的基因组可被获得,使得人类肠道微生物产生孢子的研究成为可能。几项研究建议使用孢子制剂作为传统粪便微生物移植(FMT)的替代来治疗艰难梭菌感染和IBD,由于孢子纯化过程通过与乙醇混合选择性消灭细菌、真菌和病毒,比传统的FMT更安全。此外,孢子制剂可以在受体中保持萌发和复制的活性。通过基因预测,我们发现人类肠道毛螺菌科的大多数成员都能够形成孢子。然而,还需要进行额外的实验来验证毛螺菌科孢子的实际孢子萌发潜力和安全性,以用于治疗干预。我们的研究还发现定植在动物瘤胃和人类口腔的毛螺菌科物种中不存在孢子形成相关基因,表明这些细菌占据了独特的生态位,并且可能反映了它们在各自环境中面临的选择压力和特定要求。此外,孢子形成与微生物的传播策略密切相关。
我们的研究表明,培养的毛螺菌科基因组集合增强了疾病相关基因组的分辨率,并为选择潜在有效的干预菌株提供了基础。毛螺菌科是人类肠道微生物组中的一个重要细菌家族,由于其在维持肠道稳态中的作用及其对宿主健康的影响而受到越来越多的关注。我们发现对照组中富集的大多数菌株都是未知物种,进一步强调了新物种的重要性。此外,与使用现有数据库相比,使用培养的基因组作为参考可以识别特定已知菌株的标记,从而能够进行后续的功能验证。观察到的毛螺菌科丰度与ACVD和IBD患病率之间的相关性表明,这些细菌可能对这些疾病发挥保护作用。虽然需要进一步研究来确定因果关系并阐明潜在机制,但我们的研究结果提供了有希望的见解。
代码和数据可用性
本研究中使用的所有基因组均可在公共数据库中获得,表S1提供了登录号和参考文献。对于本研究中使用的所有公共宏基因组数据,方法部分提供了网络链接或参考文献。使用的数据和脚本保存在GitHub (https://github.com/Linxiaoqianv/Lachnospiraceae_iMeta)中。补充材料(图形、表格、图形摘要和源数据)可以在在线DOI或iMeta Science (http://www.imeta.science/)中获得。
引文格式:
Xiaoqian Lin, Tongyuan Hu, Zhinan Wu, Lingne Li, Yuhao Wang, Dingyang Wen, Xudong Liu, Wenxi Li, Hewei Liang, Xin Jin, Xun Xu, Jian Wang, Huanming Yang, Karsten Kristiansen, Liang Xiao, and Yuanqiang Zou. 2024. Isolation of potentially novel species expands the genomic and functional diversity of Lachnospiraceae. iMeta e174. https://doi.org/10.1002/imt2.174
作者简介

林晓倩(第一作者)
● 华南理工大学-华大生命科学研究院联合培养生物学硕士毕业生。
● 研究方向为人体共生微生物基因组及益生菌功能研究。相关学术成果已发表于Nature Communications、iMeta等期刊。

邹远强(通讯作者)
● 博士,深圳华大生命科学研究院副研究员,中国科学院大学华大教育中心研究生导师,郑州大学硕士生导师,深圳市人体微生物检测与干预工程实验室副主任。
● 主要从事人体微生物培养组和基因组研究和益生菌的功能开发和产业转化。已在Nature biotechnology,Nature genetics,Nature Communications,iMeta等期刊发表论文30余篇,申请专利30余项。

Karsten Kristiansen(通讯作者)
● 丹麦自然科学院院士,哥本哈根大学生物学系分子生物实验室主任、丹麦自然科学院成员,曾任职丹麦农业科学院院长、挪威海洋食品和营养国家科学研究院联席主席。
● 致力于破译宿主-微生物组相互作用基础及功能微生物组挖掘与利用。在Nature、Science、Cell等顶级期刊发表论文400余篇,成果被学界广泛引用(H-index WoS,87;Google Scholar,102;总被引次数78,159),是Nature、Cell等30多家国际权威科学杂志的审稿人。

肖亮(通讯作者)
● 博士,正高级研究员,深圳华大生命科学院宏基因组平台负责人,深圳市人体共生微生物检测与干预工程实验室主任,华南农业大学兼职教授,西北大学(西安)兼职教授,中国科学院大学博士生导师,郑州大学硕士生导师,深圳市后备级人才,国家自然科学基金委函评专家。
● 以第一作者,通讯作者及共同作者身份,在Nature Biotechnology,Nature Microbiology,Nature Medicine,Cell Host & Microbe等期刊发表多篇科研论文,总计引用次数3000余次。作为项目负责人主持国自然面上项目一项及深圳市百万级技术开发项目一项,参与国家、省市级科研项目多项。现主要从事人体健康相关宏基因组学方面的科学研究与干预产品研发工作,已申请相关发明专利30余项,已获批5项,作为主要完成人完成1项相关国家标准的制定。
更多推荐
(▼ 点击跳转)
iMeta | 引用7000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期

3卷1期

2卷2期封底

2卷4期封底

3卷1期封底
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:office@imeta.science

















 1015
1015

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









