






点击蓝字 关注我们
鸭肠道微生物参考基因组及与肠道区域、发育阶段和饲养条件相关的菌群特征

iMeta主页:http://www.imeta.science
研究论文
● 原文链接DOI: https://doi.org/10.1002/imt2.198
● 2024年5月14日,浙江省农业科学院杨华、肖英平、卢立志团队在iMeta在线发表了题为 “Duck gut metagenome reveals the microbiome signatures linked to intestinal regional, temporal development, and rearing condition” 的研究文章。
● 本研究结果拓展了对鸭肠道微生物在肠段特定区域的分类学和功能上的理解,揭示了其在不同生长阶段的发展情况,以及受饲养条件影响的微生物组变化,突显了肠道微生物在家禽健康和生产中的重要作用。
● 第一作者:马灵燕、吕文涛
● 通讯作者:卢立志(lulizhibox@163.com)、杨华(yanghua@zaas.ac.cn)、肖英平(xiaoyp@zaas.ac.cn)
● 合作作者:曾涛、汪雯、赵江潮、张国龙
● 主要单位:浙江省农业科学院、阿肯色大学、俄克拉荷马州立大学
亮 点
● 首次构建鸭肠道微生物组的基因集和宏基因装配基因组;
● 鸭不同肠段微生物在分类及功能上展示了共性与特异性差异;
● 鸭肠道微生物随着生长发育多样性增加,稳定性增强;
● 水养模式下鸭肠道微生物脂多糖合成功能增强,病原菌和抗生素抗性基因丰度较高。
摘 要
鸭肠道中栖息着大量微生物,与宿主免疫、营养以及其他生命活动紧密相关,在维持鸭健康和高效生产中扮演重要角色。在本研究中,我们收集了375个肠道内容物样本,涵盖了4个不同鸭种(野鸭、麻鸭、北京鸭、番鸭)、5个不同生长阶段(3、7、14、42和70日龄)、5个不同肠段(十二指肠、空肠、回肠、盲肠、结肠)以及2种不同养殖模式(水养、旱养)。首先,构建了首个鸭肠道微生物基因目录,包含约2400万个基因,并组装了4437个宏基因装配基因组(MAGs)。分析了不同肠段微生物的分类学特征和功能,并跟踪了肠道微生物在鸭生长发育过程中的演替变化。宏基因组分析揭示了前肠和后肠微生物群落特征,以及不同肠段之间微生物群落及功能的差异。鸭肠道微生物随着生长发育多样性增加,稳定性增强,反映出宿主成熟过程中肠道微生物群落的逐渐稳定和成熟。此外,通过比较不同养殖条件对鸭盲肠微生物群落及其功能的影响,发现在水养模式下,鸭肠道微生物的脂多糖生物合成能力增强,病原菌和抗生素抗性基因丰度增加。综上所述,我们的研究结果拓展了对鸭肠道微生物在肠段特定区域的分类学和功能上的理解,揭示了其在不同生长阶段的发展情况,以及受饲养条件影响的微生物组变化,突显了肠道微生物在家禽健康和生产中的重要作用。
视频解读
Bilibili:https://www.bilibili.com/video/BV1qb421q7t2/
Youtube:https://youtu.be/sSKLjcn1sts
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
中国家禽产业占到全球家禽总量的约四分之一,其中鸭肉作为人类饮食中的重要肉类资源之一,消费量颇为可观。鸭肠道内的微生物群落在促进营养物质的消化、免疫系统发展以及提高饲料转化效率等方面发挥了至关重要的作用。多项研究证明了鸭肠道微生物受到多种因素影响,包括饮食营养成分、抗生素使用、环境温度以及微塑料的环境暴露等,凸显了其在维护健康和预防疾病中的关键作用。当前,微生物组的研究不仅有助于优化饲养管理策略,更为家禽疾病的预防和治疗策略设计提供了重要参考。
鸭的肠道是一个复杂且富于多样性的系统,孕育着不断变化的微生物群落。虽然之前的研究主要关注于盲肠或粪便中的微生物,但对整个鸭肠道微生物群落的空间分布和功能潜力仍有待深入研究。此外,尽管目前的研究已开始揭示鸡肠道微生物群随时间发展的动态变化,但鸭肠道微生物群相似研究的方法学大多局限于16S rRNA测序技术,存在着明显的局限性。因此,迫切需要利用深度宏基因组学分析方法和大规模样本的相关研究,揭示鸭微生物组的生长相关模式及功能发展特征,以期为动物健康管理提供更精确的科学依据。
在本项研究中,我们采集了375份鸭肠道内容物样本,涵盖了四个不同品种(野鸭、麻鸭、北京鸭、番鸭),5个不同生长阶段(3、7、14、42和70日龄)、5个不同肠段(十二指肠、空肠、回肠、盲肠、结肠)以及2种不同养殖模式(水养、旱养)。我们构建了一个相对全面的鸭肠道微生物基因目录(RGMGC),共含24,602,722个非冗余基因,并且重建了4,437个MAGs。宏基因组分析结果不仅揭示了鸭肠道微生物组的区域特异性和功能特定性,还追踪了鸭肠道微生物组在不同生长阶段的演变过程和成熟度。此外,我们阐明了养殖条件如何影响鸭肠道微生物群的功能特性以及对耐药基因的影响。综上所述,此项研究不但拓宽了我们对于微生物组在家禽健康养殖中的影响的认识,也为未来的肠道微生物组研究和应用提供了新的视角和数据资源。
结 果
鸭肠道微生物参考基因集和宏基因装配基因组
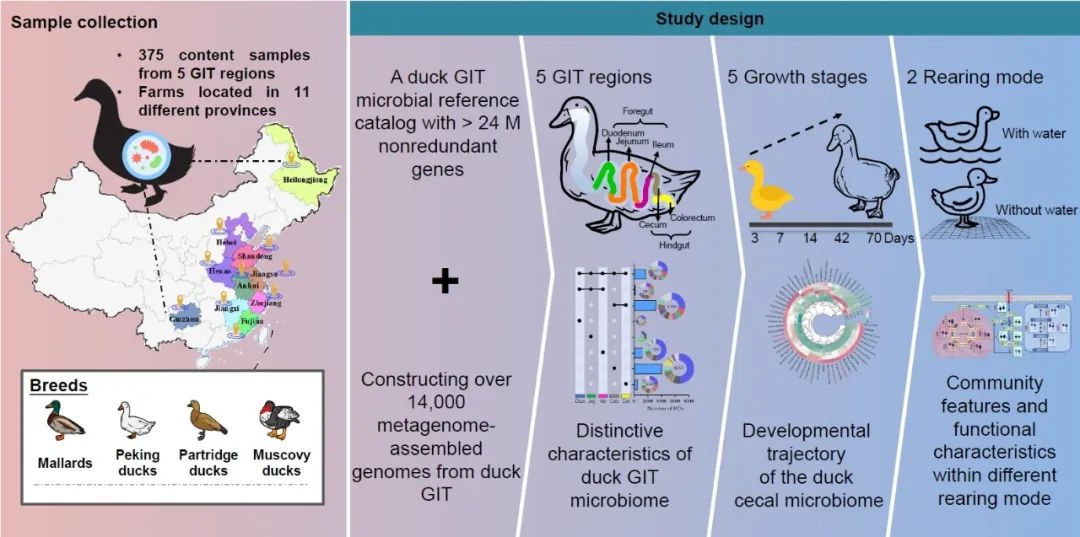
我们对从375个样本提取的基因组DNA进行了宏基因组学测序,共获得6.8 Tb的Illumina测序数据。在95%的核苷酸序列同源性聚类的基础上,构建了一个含有24,602,722个基因的非冗余鸭肠道微生物基因目录,平均基因长度为650 bp(图1A)。根据现有数据库,RGMGC中的基因按来源分类,细菌占93.2%,古菌占0.39%,真核生物占0.58%,病毒占0.21%(图1A)。此外,分别有71.65%、41.88%、3.6%的基因被注释为同源蛋白类群(COGs)、KEGG同源蛋白组(KOs)和碳水化合物活性酶组(CAZymes)(图1B)。
我们在四个品种间对基因目录进行比较分析,揭示了所展示基因数量的显著差异,顺序为:麻鸭 > 北京鸭 > 番鸭 > 野鸭。通过成对基因重叠分析,我们发现约60%的基因是共有的,麻鸭为39.90%,北京鸭为37.80%,番鸭为31.79%,野鸭为26.50%(图1C)。随后通过Shannon指数和Beta多样性评估每个品种基因、属和KOs的相对丰度(图1D)。
进一步将鸭的基因目录与人类(Homo sapiens,9.9 M)、猪(Sus scrofa domesticus,7.7 M)和鸡(Gallus gallus domesticus,9.04 M)的基因目录进行了比较。通过在基因序列水平上进行成对叠加分析,我们观察到每个物种都拥有大量独特的基因,超过70%的基因是种内独有的(鸭92.12%,猪72.51%,鸡76.61%,人78.62%)。相对的,所有这些物种共有的基因仅占很小比例(约0.9%)(图1E)。此外,在属及功能水平上,鸭肠道菌群的Alpha和Beta多样性指数均高于其他物种(图1F)。
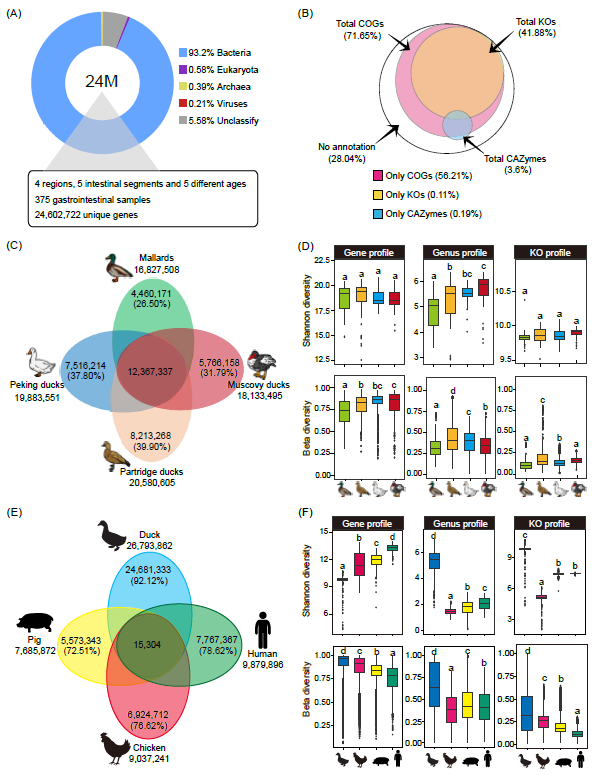
图1. 鸭肠道微生物组基因参考目录构建
(A)鸭肠道微生物基因参考目录分类学注解分析;(B)根据三个功能类别(COGs、KOs和CAZymes)的注解对RGMGC进行标注。显示在指定功能类别中鉴定到的基因百分比;(C)维恩图用以展示不同鸭品种基因目录中独有及共有基因的分布;(D)四个鸭品种在基因、属以及KO功能水平上的Shannon指数和Beta多样性;(E)鸭、鸡、猪和人基因目录中的重叠和独特基因;(F)鸭、猪、鸡和人的基因、属以及KO功能水平上的Shannon指数和Beta多样性。
我们进一步研究了基于指定MAGs的主要细菌门类的分布(图2A)。最丰富的门类是Firmicutes_A(n = 1920),主要由Bacteroidia和Clostridia类组成,其次是Bacteroidota(n = 1089)。此外,Firmicutes(n = 310)、Proteobacteria(n = 212)、Actinobacteriota(n = 152)和Desulfobacterota(n = 119)占比较高。在考虑古生菌门时,主要群体是Methanobacteriota(n = 14)、Halobacteriota(n = 15)和Thermoplasmatota(n = 5)。在MAGs中,约99.23%能在细菌门级别进行注释,而0.77%在古生菌进行注释。在属级别,87.02%的MAGs被划分到菌属水平,其中99.12%属于细菌领域,剩下的0.88%划归到古生菌属。值得注意的是,相当一部分MAGs(33.25%)被进一步分类为物种级基因组bins(SGBs),包括98.85%的细菌物种和1.15%的古生菌物种(图2B)。
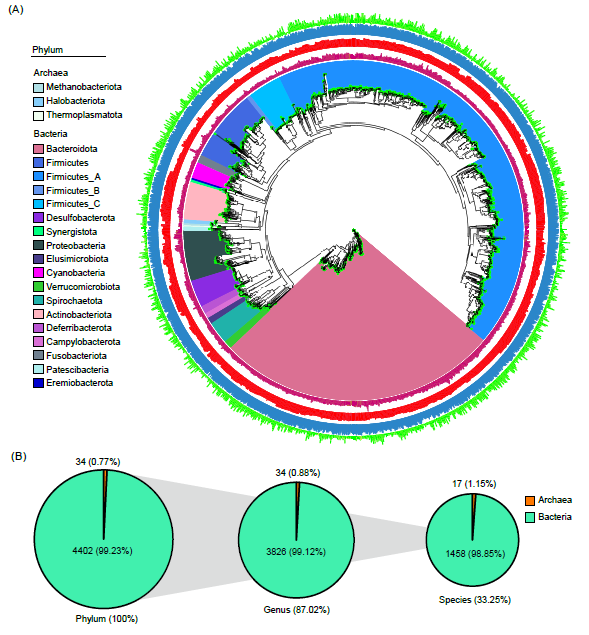
图2. 鸭类中基因组组装细菌(MAGs)的生成与质量评估
(A)使用PhyloPhlAn构建的系统树,展示了从鸭肠道(GIT)各个区域采样得到的4437个MAGs。各系统进化支按门类进行着色;(B)在门、属、种级别上的基因组比例及具体数量。
鸭肠道不同区域微生物群落和功能的分类特征
为探索鸭不同肠段的微生物组特征,我们采集了不同肠段样本,包括小肠的十二指肠、空肠和回肠,以及大肠的盲肠和结肠样本。鸭类肠道按形态和功能差异可分为前肠(十二指肠、空肠和回肠)和后肠(盲肠和结直肠)(图3A)。
前肠各部位的微生物多样性相似,自十二指肠至回肠呈现轻微增长趋势(图3B)。在后肠各部位中观察到了类似的模式(图3B)。然而,Alpha多样性和Beta多样性的指标显示前肠的微生物多样性高于后肠(图3B)。主成分分析(PCoA)确认前肠和后肠部位之间存在明显的分离(图3C)。此外,进一步针对GIT五个区域的微生物群落分析显示出差异显著。例如,Firmicutes在前肠部位更为丰富,而Bacteroidetes在盲肠和结直肠区域显示出高度富集。特定的微生物类群在特定的肠段区域内表现出显著富集模式。例如,Helicobacter和Prevotella主要集中在十二指肠;Lactobacillus和Enterococcus在回肠更为富集;大肠中Bacteroides、Alistipes、Clostridium和Blautia丰度更高;而Escherichia在小肠中相对含量较高(图3D)。
进一步分析不同肠段肠道菌群功能差异,通过横向对比肠道各区域的功能模块,如KOs,我们更深地理解了肠道微生物群的区域功能差异性(图3F)。前肠段微生物功能主要富集辅因子、维生素及氨基酸的代谢过程等功能,相较之下,后肠段微生物则脂质代谢以及蛋白质折叠、分选和降解功能更为显著(图3F)。
图3. 鸭肠道微生物的功能与分类学特征
(A)鸭肠道示意图;(B)鸭肠道微生物在基因、属和KO功能水平的Alpha多样性和Beta多样性;(C)基于Bray-Curtis距离的GIT分类学和(D)功能的PCoA分析;(E)肠道不同区域中占主导微生物属水平的相对丰度;(F)鸭肠道各区域微生物组功能变化。
鸭肠道微生物碳水化合物活性酶的区域特征
CAZymes是肠道中代谢复杂碳水化合物最重要的酶。我们在前肠微生物中发现了更高丰度的碳水化合物酯酶(CEs)和糖基转移酶(GTs),表明它们具有催化取代糖类和糖基转移的能力。相比之下,前肠微生物展示出更多比例的糖苷水解酶(GHs),表明它们具有更强的水解碳水化合物中糖苷键的能力(图4A)。
超过30,000个CAZymes由4,437个MAGs编码,其中最常见的类别是GHs,其次是GTs和CBMs(图4B)。Firmicutes和Bacteroidetes,以及Proteobacteria和Actinobacteria,分别展示了最大和最多样化的CAZymes库(图4C)。Firmicutes和Bacteroidetes,显示了高比例的GHs,而Clostridia显示了更高比例的AAs(图4C)。我们还比较了在五个肠段富集的MAGs的丰度,基于LEfSe分析鉴定了共计60个MAGs(LDA > 3;p < 0.05)(图4D)。例如,前肠特别富含十二指肠和空肠相关的MAGs,主要属于Lachnospiraceae、Ruminococcaceae和Oscillospiraceae科(图4D)。此外,在回肠部分,Butyricioccaceae、Burkholdriaceae、Acutalibacteraceae、Borkfalkiaceae和Bacteroidaceae是最丰富的MAGs分类家族(图4D)。对于后肠,梭菌科在盲肠中占比较大,而Atopobiaceae、Desulfovibrionaceae、Enterococcaceae和Fusobacteriaceae则在结肠中发现。在前肠道,微生物群主要由GHs、GTs和CEs主导;对于后肠,CBMs的比例较高,CEs的比例下降(图4D)。这些结果表明,在不同的肠段中,不同的菌群物种编码了相似的CAZymes。
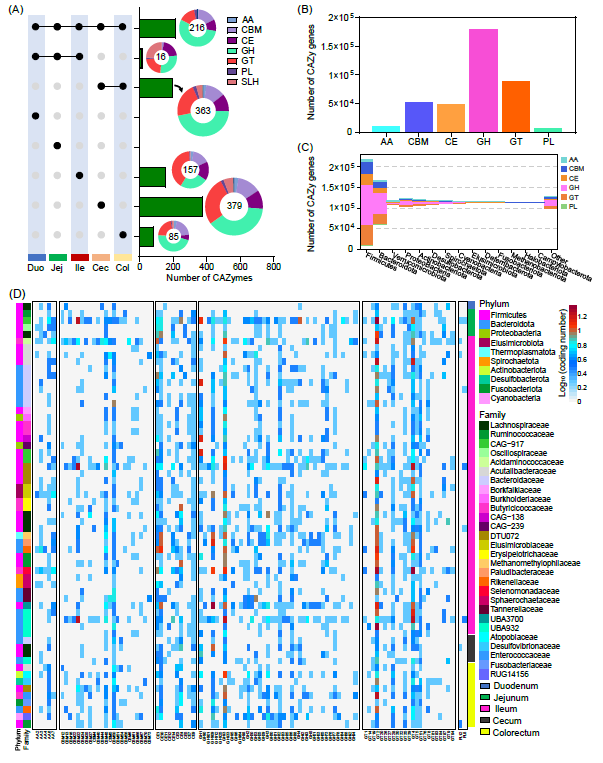
图4. 鸭肠道微生物组碳水化合物活性酶的区域特征
(A)鸭各肠道微生物组中CAZymes比较分析;(B)基于MAGs的CAZymes基因数量;(C)按主要门水平分类的基因组中CAZyme家族的分布;(D)热图显示了基于LEfSe分析得到的60个特征差异CAZymes。
鸭肠道微生物随生长阶段的发育轨迹和演替变化
随着鸭的成长,微生物多样性呈增加趋势,在第14天达到基因和属水平的高峰,此后保持稳定或略有下降(图5A,B)。值得注意的是,对于麻鸭(MD)和北京鸭(PD),PCoA散点图根据不同阶段组明显聚类(图5C)。特别是42天和70天的群组显示出较高的相似性,表明在晚期肠道微生物群落稳定(图5C)。相比之下,3日龄的肠道微生物组显示出显著的差异,这一观察结果可归因于短期内对外部环境的暴露以及在这一早期发展阶段微生物群落的初步建立(图5C)。
在整个生长阶段中,无论是MD还是PD品种,它们的肠道微生物菌群中占主导地位的门类均为Firmicutes、Proteobacteria、Bacteroidetes、Fusobacteria和Actinobacteria。其中,数量最为丰富的Firmicutes菌群数量从第3天开始稳步增加,直至第70天。而Proteobacteria从第3天开始下降,到第14天达到最低点,随后又逐渐回升,直到第70天(图5D)。在3日龄的时候,鉴定出的主要属包括Klebsiella、Lactobacillus、Escherichia和Enterobacter(图5E)。到7日龄时,Clostridium、Enterococcus和Lactococcus的丰度更为丰富(图5E)。在中期生长阶段,Blautia、Eubacterium和Bacteroides的丰度逐渐丰富(图5E)。在后期生长阶段,Lactobacillus、Roseburia、Butyrivibrio和Bifidobacterium的丰度增加(图5E)。且微生物组功能在第7日龄达到峰值,此后保持稳定(图5F)。
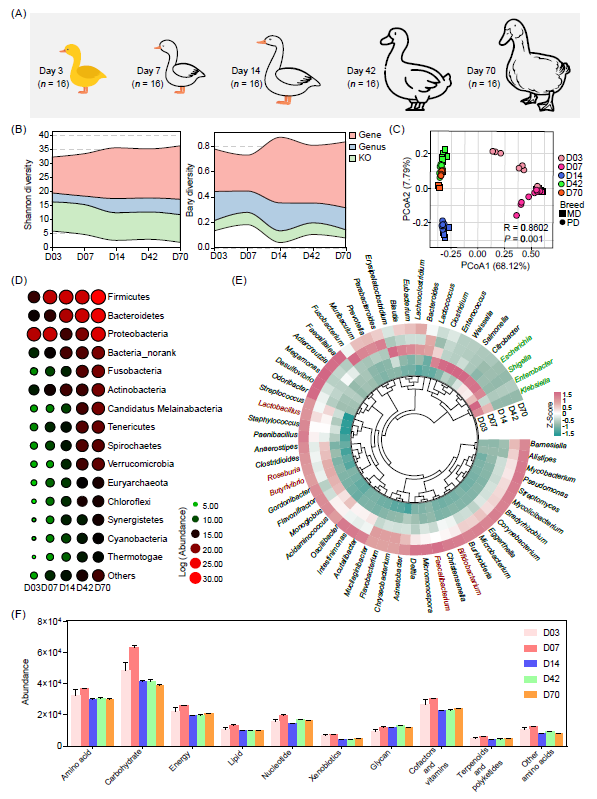
图5. 鸭肠道微生物随生长阶段的发育轨迹和演替变化
(A)鸭生长阶段示意图;(B)不同生长阶段肠道微生物在基因、属和KO功能水平的Shannon指数和Beta多样性;(C)基于Bray-Curtis距离的分类PCoA分析;(D)不同生长阶段主要门类的相对丰度;(E)不同生长阶段主要属的相对丰度;(F)不同生长阶段肠道微生物的功能丰度变化。
不同养殖条件下鸭肠道微生物组成及功能特征
与旱养模式(WOW)条件相比,水养模式下(WW)增加了属水平和功能KOs的Alpha多样性,并降低了基因的Beta多样性(图6A)。PCoA分析显示了不同养殖条件下的明显聚类。此外,在WOW条件下,Firmicutes和Bacteroidetes的平均相对丰度分别增加和减少。
通过在物种水平上比较WW和WOW下鸭肠道菌群变化,结果揭示了不同养殖组间的富集模式(图6B)。共有77个MAGs聚类成36个种,显示出差异性模式。例如,属于18个SGB中的39个MAGs在WOW养殖鸭中富集,包括链球菌属的Streptococcus alactolyticus、Bilophila wadsworthia和拟杆菌属的Bacteroides eggerthii,而剩余的38个MAGs聚类为18个SGB在WW养殖鸭中富集,大多属于Phocaeicola plebeius_A。更重要的是,我们发现在WOW养殖鸭中,阿克曼氏菌Akkermansia muciniphila的丰度更高(图6B)。
我们进一步比较了WW和WOW条件下肠道菌群的功能差异。WOW条件在涉及到碳水化合物、氨基酸、脂质、能量代谢的途径中表现出显著富集。相反,WW条件影响了辅因子和维生素代谢、多糖代谢、萜烯和多酮代谢以及核苷酸代谢等途径(图6C,D)。KEGG分析进一步证实,WOW条件下鸭肠道菌群氨基酸生物合成以及淀粉和蔗糖的代谢增加,而在WW条件下,则富集了涉及脂多糖生物合成、TCA循环、多糖降解和抗生素生物合成的菌群功能。进一步研究发现,WW条件下富集的KEGG途径主要涉及酶的参与(图6E)。在WW条件下,观察到蔗糖磷酸转移酶基因(scrA)的减少,同时蔗糖酶基因(sacA)和葡萄糖-6-磷酸异构酶(GPI)的丰度增加(图6F)。
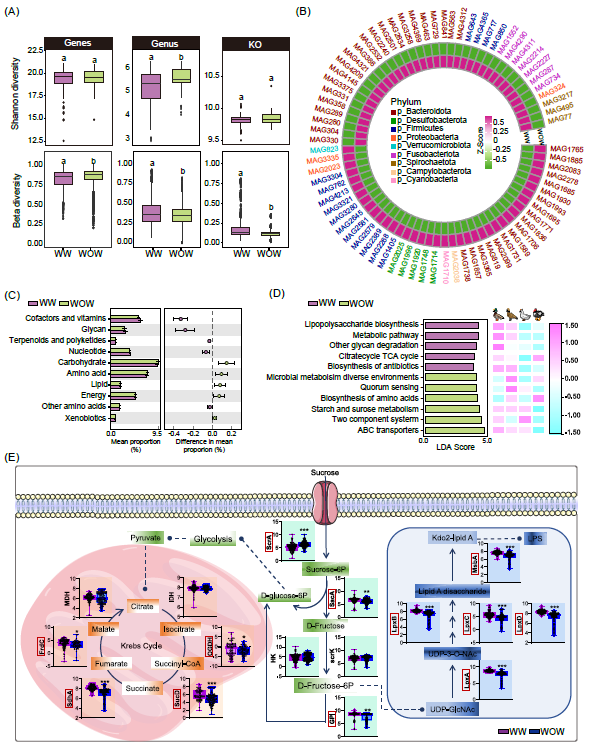
图6. 不同养殖条件下观察到的功能性变化
(A)在不同养殖条件下肠道微生物在基因、属和KO功能水平的Alpha多样性和Beta多样性;(B)热图展示了在不同养殖条件下特定MAGs的富集情况;(C)在不同养殖条件下肠道微生物组主要KEGG功能差异;(D)不同养殖条件下鸭微生物组KEGG的LEfSe分析。(E)不同养殖模式鸭肠道菌群功能基因差异。
不同养殖条件下鸭肠道耐药基因变化及分布特征
与WOW养殖模式下相比,WW养殖模式下鸭肠道ARGs在Beta多样性上显示出显著差异,且ARGs的Shannon指数略有增加(图7A,B)。与WOW组相比,WW组中多重耐药类、四环素类、β-内酰胺类、卡邦酰胺类、氯霉素类、磺胺类、甲胺蝶呤类、春雷霉素类和甲氧苄啶类ARGs丰度高,而万古霉素类、杆菌肽类、萎蔫酸、M-L-S类(大环内酯-林可霉素-链霉素类)、氨基糖苷类、普鲁霉素类和磷霉素类ARGs丰度低(图7B)。普氏分析揭示了细菌群落与ARGs组成之间的显著相关性(图7C)。Actinobacteria、Bacteroidota、Firmicutes和Proteobacteria菌门主要负责CARD编码(图7D)。WOW和WW组中富集的227个MAGs的系统发育树显示,WW养殖模式下鸭肠道菌群富集的MAGs主要属于Bacterioidota和Proteobacteria,这些门主要编码氟嘧啶、大环内酯、糖肽和四环素类抗生素(图7E)。
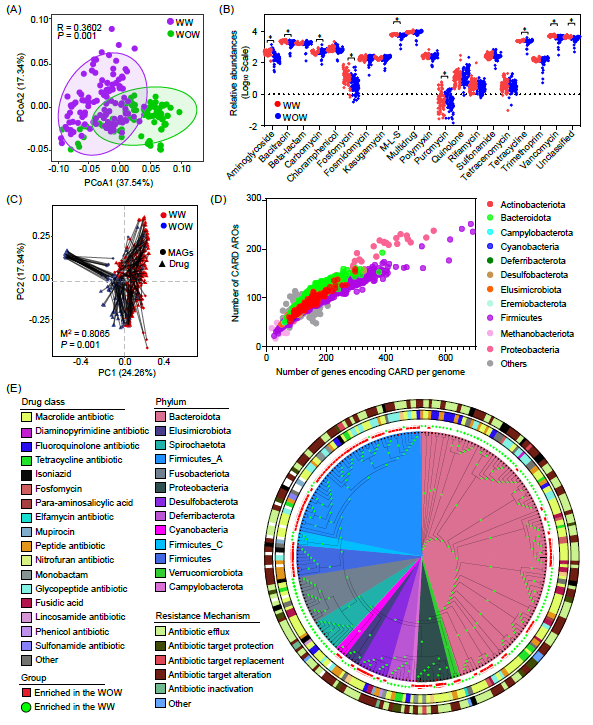
图7. 不同养殖条件下的鸭肠道抗生素抗性基因特征
(A)基于Bray-Curtis距离的ARGs的PCoA分析;(B)抗生素类别的相对丰度变化;(C)普氏分析显示了MAGs组成和抗生素类别之间的相关性;(D)基因组中编码抗生素抗性基因的数量;(E)在不同养殖条件下富集的MAGs的系统发育树。
讨 论
鸭不同肠段肠道微生物的结构功能特征
鸭肠道微生物组呈现出显著的区域特异性和功能多样性,对禽类的生产和健康至关重要。本研究通过对鸭不同肠段的菌群多样性进行深入分析,揭示了各肠段的微生物组成及功能变化。小肠中的微生物群落以乳酸菌为主导,特别是Lactobacillus、Enterococcus和Lactobacillus等,它们在鸭健康和营养吸收过程中发挥着核心作用,促进了复杂碳水化合物的发酵过程,并有助于短链脂肪酸的产生,进一步调节宿主的能量代谢和免疫功能。相比之下,大肠中的微生物群落以专门降解黏蛋白和高级碳水化合物的种类为特征,如Bacteroides和Alistipes。这种功能的存在强调了宿主对膳食纤维的降解和利用的依赖性,以及肠道微生物在这一代谢活动中的参与程度。Ruminococcus属在盲肠区域的高度丰富性,可能预示了其在抗炎反应和维持肠道屏障功能中的重要角色。其中,Ruminococcus与丁酸盐的产生相关,并有可能通过对复杂碳水化合物,如果胶和多糖的代谢,来维持宿主的消化健康。此外,本研究探讨了鸭肠道微生物组编码的多种CAZyme家族,这些酶具有促进多样化碳水化合物基质在肠道中的有效利用的作用。此类酶类在前肠与饮食成分紧密相关,而后肠主要是发酵和吸收功能,其中CAZyme家族表现出高度的稳定性和普适性。这一发现可能对于理解鸭在消化过程中对特定营养素的特殊代谢需求及它们如何影响宿主的整体健康具有深远意义。
鸭肠道微生物随生长阶段的发展轨迹
肠道菌群的成熟过程揭示了不同生命阶段中微生物定植的动态进程。早期研究指出雏鸡的肠道菌群与成年鸡相比存在明显差异。与此相一致,我们的研究发现在鸭肠道菌群在3-7日龄时有显著变化,其后逐渐向多样性提升和稳定性增强的方向发展。这一模式与人类幼年时期肠道菌群的变化类似,显示出跨物种的微生物动态共性。PCoA结果表明,相较于品种,鸭盲肠菌群更依据年龄呈现分类群组,指示出一种按生长阶段发展的生物过程。这些综合性发现表明,随着鸭宿主的成长,肠道微生物群落演变为一相对成熟且稳定的体系。前期研究指出,在北京鸭早期盲肠微生物组发展中,变形菌门占主导地位。本研究亦在第三天观察到变形菌门的显著富集,包括一些已知的病原菌如Klebsiella pneumoniae、Escherichia coli等,在生命初期阶段较为常见,这反映了肠道微生物群体环境短期接触的初期动态。反之,在鸭的中期生长阶段,产生丁酸的益生菌Agathobaculum数量上升,表明了菌群向更加稳定、健康、成熟的方向转变。其他如Lactobacillus和Butyrivibrio等关键的丁酸生产菌在生长阶段中期持续出现,暗示着菌群环境正在向更优的状态过渡,这可能对鸭的生长性能和疾病抵抗力产生积极影响。此外,在鸭成长的不同阶段,微生物群落中特定代谢途径的丰富化,反映了宿主发育需求与微生物群落的代谢能力之间的动态互作。
不同养殖模式对鸭肠道微生物和抗性基因的影响
饲养环境对家禽的性能及其肠道微生物多样性具有显著影响。随着中国鸭肉产业朝着高效集约化方向发展,本研究着重分析旱养与水养条件下鸭盲肠微生物组的差异。研究表明,旱养条件下鸭肠道菌群显示出较高的Alpha多样性。在旱养鸭群体中,Bacteroides属的显著富集揭示了其在膳食及内源性多糖降解方面的强大能力。此外,WOW组鸭肠道菌群中A. muciniphila的增加强调了其在维持肠道屏障完整性和调节免疫反应中的重要作用。另一方面,水养条件促进了Paraprevotella和Phocaeicola的增长,而Fusobacterium的富集可能与感染风险增加相关,尤其是对幼鸭而言。长期暴露于过量内毒素LPS可能导致炎症反应、肠道屏障功能障碍,并增加宿主对感染及炎症性疾病的易感性。水养模式下,由于肠道中关键酶lpxB、lpxC和lpxD表达增加,鸭肠道菌群LPS生产能力得到加强。这一现象表明,鸭肠道微生物在水养条件下通过协同调节lpx基因簇酶实现了LPS生物合成的调节机制。此外,与水养条件相关的肠道微生物群促进了TCA循环的相关功能,反映了对有效糖代谢的适应,以满足生长和代谢需求。
宏基因组测序结果显示,水养模式下鸭肠道微生物中耐药基因的Shannon指数略有提高,揭示了水养条件可能影响肠道微生物群中ARGs的多样性和丰富度。水养系统可能增加了抗性基因通过环境抗生素抗性细菌储库传播到家禽种群的可能性。我们在WW组鸭肠道微生物中观察到多重耐药类、四环素类和β-内酰胺类等主要抗生素类别ARGs的富集,暗示了水体环境在家禽中传播抗生素抗性的关键作用。以上对理解养殖条件、肠道微生物功能与鸭肠道ARGs的存在及其相互关系提供了深入见解,有助于优化家禽业的饲养实践和健康管理策略。
方 法
实验设计与样本收集
对于宏基因组测序分析,总共收集了375份肠道样本,这些样本来自中国的不同省份,包括黑龙江、浙江、江西、河南、上海、贵州、山东、江苏、安徽、福建,和河北等不同养殖地的鸭群。该研究设计分为以下四部分:(1)375个胃肠道样本,包括四个品种(野鸭,番鸭,北京鸭和麻鸭);五个肠段(前肠部分的十二指肠,空肠,和回肠;以及后肠部分的盲肠和结肠);以及五个不同生长阶段(3,7,14,42和70日龄)。这些样本用于建立鸭肠道微生物参考基因集;(2)选取了来自北京鸭(PD,n = 12/肠段)和麻鸭(MD,n = 12/肠段)的肠道样本用于研究鸭肠道宏基因组的肠道区域特征(总样本数为120);(3)选取北京鸭(PD,n = 8/日龄)和麻鸭(MD,n = 8/日龄)的五个不同日龄段(3,7,14,42和70日龄)的盲肠样本用于分析研究肠道微生物随生长阶段的变化;(4)选取野鸭(n = 30),番鸭(n = 62),北京鸭(n = 51)和麻鸭(n = 62)的盲肠内容物样本,根据饲养条件将其分为两组:旱养模式组(WW,n = 91)和水养模式组(WOW,n = 114),用于探究不同养殖模式对鸭肠道菌群和抗生素抗性基因的影响。
鸭肠道微生物参考基因集构建
从375个样本中,测序产生了总共6.8 Tb的Illumina数据,大约45.7亿次测序读取,读取长度为150 bp。首先使用Trimmomatic(v.0.33)修整了Illumina数据的接头。之后,为了降低潜在的环境DNA污染,使用BWA-MEM(v.0.7.17)将序列数据映射到宿主和人类基因组上。剩余的共计1.7Tb的测序读取被视为高质量读取,其中包括野鸭的338.7 GB,麻鸭的330.4 GB,番鸭的322.5 GB以及北京鸭的313.6 GB;3日龄为15.5 GB,7日龄为48.1 GB,14日龄为44.5 GB,42日龄为69.6 GB,70日龄为59.1 GB。
宏基因组装分析
在每个样本的连续序列片段上进行了宏基因组的装箱分析操作。利用MetaBAT2、MaxBin和CONCOCT软件将高质量的连续序列片段进行装箱分析,获得了多个MAGs。为了提升MAGs的组装质量,采用MetaSPAdes软件对MAGs进行重新组装,并通过bwa mem方法从洁净数据中提取所需信息。经过质量控制及数据过滤后,共有4,437个MAGs的质量达到中等以上(完整性 ≥ 50%,污染度 ≤ 10%)。进一步采用metaWRAP quant_bins模块计算每个MAGs的丰富度,计算过程基于TPM法计算标准。根据Nayfach等人的算法,根据完整性和污染度校正了估计的基因组大小。随后,将所有装箱中的基因转换为蛋白质序列,从而得到了每个装箱的蛋白质组。这些蛋白质组后续用于Phylophlan重建系统发育树,并利用Evolview(v.3)及iTol(v.4.3.1)进行可视化。基因组使用GTDB-Tk(v.0.1.6)进行注释,该过程基于基因组分类数据库。
统计分析
采用Shannon指数衡量属、基因和KOs的多样性。基于Bray-Curtis评估细菌群落结构和功能概况的整体差异,并通过PERMANOVA和ANOSIM测试来评估各组之间的差异,最终通过PCoA图进行可视化展示。LEfSe分析用以识别在特征(属、MAGs、功能)之间的显著差异。元基因组谱统计分析(STAMP)用于比较来自不同养殖条件的微生物群落功能差异。使用TBtool软件构建热图。
代码和数据可用性
宏基因组测序数据已提交至 NCBI 序列读取存档 (SRA) 数据库,研究登录号为 PRJNA1064443 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1064443) 和 PRJNA871932 (https:// www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA871932)。使用的数据保存在GitHub(https://github.com/Lingyma/Maly2024-iMETA)中。所有的补充材料(文本、图、表、中文翻译版本或视频)也可从线上获取。
引文格式:
Ma, Lingyan, Wentao Lyu, Tao Zeng, Wen Wang, Qu Chen, Jiangchao Zhao, Guolong Zhang, Lizhi Lu, Hua Yang, and Yingping Xiao. 2024. “Duck Gut Metagenome Reveals the Microbiome Signatures Linked to Intestinal Regional, Temporal Development, and Rearing Condition.” iMeta 3, e198. https://doi.org/10.1002/imt2.198
作者简介

马灵燕(第一作者)
● 浙江省农业科学院农产品质量安全与营养研究所博士后。
● 研究方向为肠道微生物与宿主代谢。以第一作者在iMeta、Gut Microbes、Molecular Nutrition and Food Research等期刊发表SCI论文10余篇。

吕文涛(第一作者)
● 浙江省农业科学院助理研究员。
● 研究方向为畜禽养殖与质量安全。以第一或通讯作者在Journal of Animal Science and Biotechnology、Poultry Sciences、Food Function等期刊发表SCI论文10余篇。

卢立志(通讯作者)
● 浙江省农业科学院畜牧兽医研究所研究员。
● 国家水禽产业技术体系遗传改良研究室主任、蛋鸭品种改良岗位专家。主要研究方向为家禽遗传育种。以第一或通讯作者在Genome Biology、Food Chemistry、Poultry Sciences等期刊等发表论文60余篇。主持国家重点国际合作专项、国家农业科技跨越计划、国家重点研发项目课题等。

杨华(通讯作者)
● 浙江省农业科学院党委副书记,研究员。
● 国家水禽产业技术体系岗位科学家,研究方向为病原微生物与细菌耐药。以第一或通讯作者在iMeta、Food Chemistry、Journal of Animal Science and Biotechnology等期刊发表学术论文40余篇。主持国家水禽产业技术体系、国家重点研发计划课题、浙江省重点研发项目等。

肖英平(通讯作者)
● 浙江省农业科学院畜产品质量安全研究室主任,研究员。
● 研究方向为肠道微生物与畜禽健康养殖。以第一或通讯作者在iMeta、Gut Microbes、Journal of Advanced Research等期刊发表学术论文40余篇。主持国家自然科学基金项目、国家重点研发子课题、浙江省农业重大科研专项课题等;以第一发明人身份授权发明专利10件;牵头制定发布标准3项。
更多推荐
(▼ 点击跳转)
iMeta | 引用7000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期

3卷1期

2卷2期封底

2卷4期封底

3卷2期
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 20)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、50万用户的社交媒体宣传等。2022年2月正式创刊发行!目前期刊已经被ESCI、Scopus等数据库收录。
联系我们
iMeta主页:
http://www.imeta.science
姊妹刊iMetaOmics主页:
http://www.imeta.science/imetaomics/
出版社iMeta主页:
https://onlinelibrary.wiley.com/journal/2770596x
出版社iMetaOmics主页:
https://onlinelibrary.wiley.com/journal/29969514
iMeta投稿:
https://wiley.atyponrex.com/journal/IMT2
iMetaOmics投稿:
https://wiley.atyponrex.com/journal/IMO2
邮箱:
office@imeta.science

















 1887
1887

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









