






点击蓝字 关注我们
美吉生物云 2024:升级单细胞转录组和多组学云流程

方法论文
● 期刊: iMeta (IF 23.7)
● 原文链接DOI: https://doi.org/10.1002/imt2.217
●2024年6月25日,上海美吉生物医药科技有限公司团队在iMeta在线发表了题为“Majorbio Cloud 2024: Update single-cell and multiomics workflows”的文章。
● 本研究在已开发的美吉生物云 (https://cloud.majorbio.com/) 在线数据平台上发布了3款单组学云流程,2款多组学流程以及流程拓展工具助力多组学数据的挖掘和意义阐释。
● 第一作者:韩畅、史彩萍、刘林梦、韩继臣
● 通讯作者:韩继臣(jichen.han@majorbio.com)、韩畅(chang.han@majorbio.com)
● 合作作者:黄华生、张祥林、喻克刚、杨倩倩、汪妍、李晓丹、傅文垚、高豪
● 主要单位:上海美吉生物医药科技有限公司
亮 点
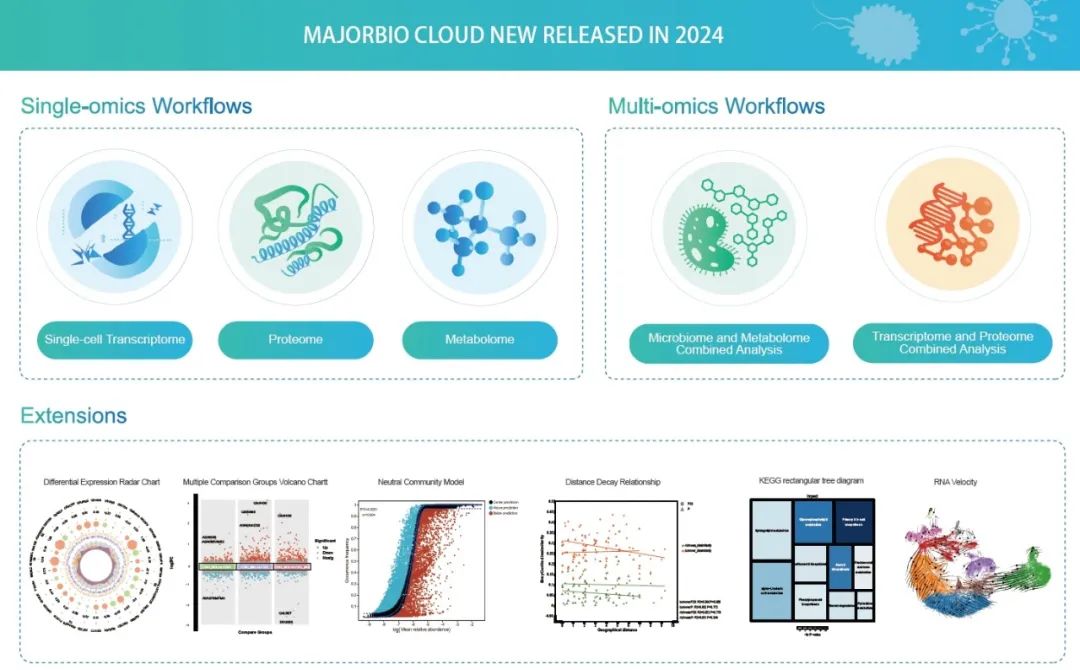
● 美吉生物云助力科研工作者分析复杂的多组学数据,降低组学数据生物学意义挖掘的门槛;
● 全新发布单细胞转录组、蛋白组、代谢组工作流,以及多组学联合分析工作流;
● 发布一种全新的交互式分析模式“工作流+拓展工具”。
摘 要
美吉生物云 (https://cloud.majorbio.com/) 是一站式在线数据分析平台,旨在促进生物信息学服务的发展,帮助科研工作者逾越干试验与湿实验之间的鸿沟,加速生命科学研究发现和成果的产出。在2024年,我们发布了3款单组学云流程,2款多组学流程以及流程拓展工具助力多组学数据的挖掘和意义阐释。
视频解读
Bilibili:https://www.bilibili.com/video/BV1ib421n7Zy/
Youtube:https://youtu.be/mJgAaWukTXI
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
高通量多组学技术的发展深刻地影响生命科学和基础医学研究,例如:利用测序和质谱技术,为不同治疗方案下的预后效果的预测性生物标志物的发现提供更多维度的可能性。前沿的多组学技术使得科研工作者可以更深入地理解疾病和健康的生物学过程和分子功能。新兴的组学策略和新型的仪器不断朝着更高的通量和更低的检测成本发展。不断增长的多组学数据需要能够访问数据资源并以简单、快速和准确的方式进行分析,因此,迫切地需要开发和应用适宜的生物信息学工具和流程来阐释这些数据。自动数据分析和数据可视化是组学数据分析的两个关键要素。
生物信息学分析平台,如Cell Ranger、MetaboAnalyst、GEPIA2和iNAP,提供了访问数据和计算结果的Web界面。但是,这些交互友好的 Web 服务是为单一类型的组学而设计的。Majorbio Cloud (https://cloud.majorbio.com/) 提供了一种简单而强大的方法来分析常规转录组、单细胞转录组、蛋白质组、代谢组、宏基因组和其他组学数据,助力研究人员分析复杂的多组学数据并推断组学数据的生物学意义。自Majorbio Cloud在iMeta上首次发表以来,吸引了全球研究人员的关注,并被有组学分析需求的研究人员广泛使用。此外,美吉生物云还是一个互动交流和组学知识传播平台。
工作流一:单细胞转录组工作流
单细胞RNA测序(scRNA-seq)是一种新兴技术,用于在单个细胞水平上对遗传物质进行高通量测序分析。它已广泛应用于免疫学、发育生物学、肿瘤学、心脏病学和神经生物学。单细胞转录组工作流程是一个用于高维单细胞转录组数据挖掘的简单易用、高效的分析流程,包括以下六个步骤:(1)数据预处理;(2)细胞过滤;(3)批量效应去除和样品合并;(4)聚类;(5)标记基因鉴定;(6)下游分析。过程如下:使用具有默认参数的 Cell Ranger (v7.1.0) 处理读取。Illumina测序仪生成的FASTQ文件与基因组对齐。使用Seurat软件包根据每个样本的UMI计数和mito %进行细胞归一化和回归,以获得标准化后的数据,并通过函数NormalizeData对其进行归一化以进行进一步分析。函数 FindVariableGenes 用于计算单个细胞中的高可变基因(HVG)。无监督细胞簇结果基于主成分分析(PCA)的前30个主成分,通过应用Seurat软件包中的基于图的聚类方法(分辨率0.8)生成。对于子聚类,我们对一组特定的数据(通常仅限于一种细胞类型)应用了相同的归一化、降维和聚类过程。对于每个簇,我们使用 Wilcoxon 秩和检验来查找比较其余Cluster的差异表达基因。SingleR和已知标记基因用于鉴定细胞类型。下游分析,如不同细胞类型的差异表达基因和通路富集、拟时序分析和细胞通讯分析,可用于揭示样品中各种类型细胞的功能、状态和相互作用(图1)。

图1. 单细胞转录组工作流
工作流包含了6步:(1)数据预处理;(2)细胞过滤;(3)批量效应去除和样品合并;(4)聚类;(5)标记基因鉴定;(6)下游分析。DEG,差异表达基因;GSEA,基因集富集分析;TF,转录因子。
工作流二:蛋白组学工作流
蛋白组学工作流程是一个用户友好、分析全面的数据挖掘流程,适用于基于数据非依赖型的采集模式 (DIA)、非标记定量 (LFQ) 和串联质量标签 (TMT) 的定量蛋白质组学数据(图 2)。蛋白质组学标准化工作流由七个主要模块组成:数据处理、蛋白质表达和功能注释、统计分析、蛋白质集分析、加权基因相关网络分析 (WGCNA)、基因集富集分析 (GSEA) 和时间序列数据分析。我们为医学队列研究提供了额外的分析模块 —— 生物标志物发现和模型构建模块。
蛋白质组学数据处理模块用于低质量数据过滤和缺失值估计。蛋白质表达和功能注释模块包括Venn、PCA、相关性分析以及基于数据库或软件的功能注释。提供了配对/非配对T检验、方差分析、Kruskal-Wallis 检验和事后检验,用于蛋白定量数据的统计学意义的检验。蛋白集是依据蛋白质表达谱、功能注释、生物通路富、用户研究背景所生成的与表型相关的蛋白质列表,用户可以生成他们感兴趣的蛋白质集,并通过聚类、蛋白质-蛋白质相互作用、通路分析、功能富集等来解释数据。Lasso-Logistic/Cox回归、随机森林和支持向量机可用于疾病风险预测、早期诊断、预后和监测。
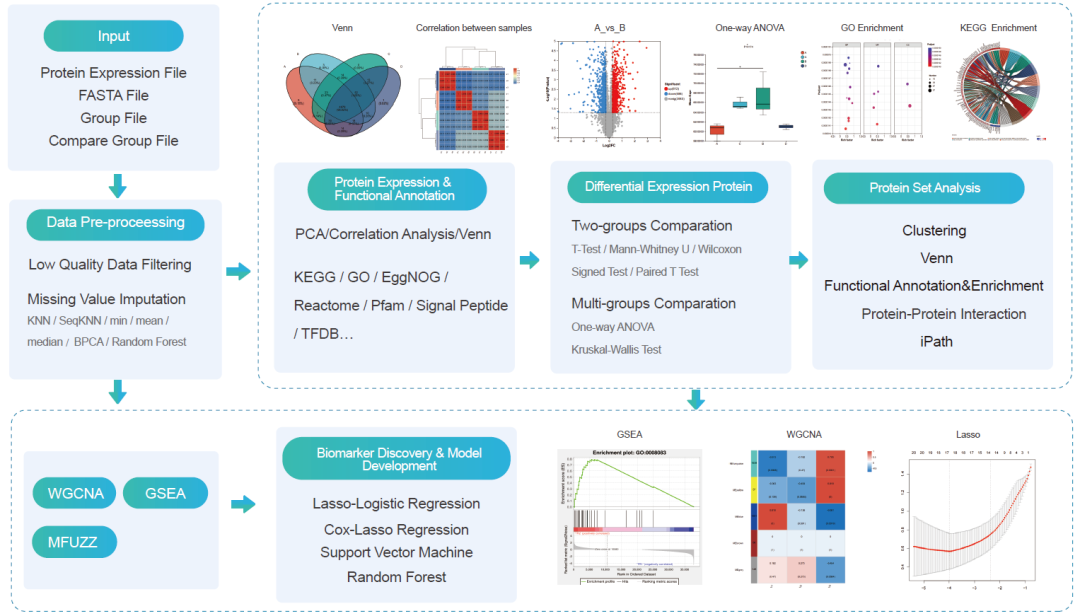
图2. 蛋白组学工作流
蛋白质组学标准化工作流由七个主要模块组成。我们为医学大队列研究项目额外提供生物标志物发现和模型构建分析模块。ANOVA,方差分析;KNN,K近邻分类;PCA,主成分分析。
工作流三:代谢组学工作流
代谢组学研究主要基于使用液/气相色谱-质谱(LC/GC-MS)和核磁共振(NMR)来检测、鉴定和定量生物体中的小分子代谢物。代谢组学数据庞大而复杂,通常需要专门的数据分析软件以及广泛的化学信息学、生物信息学和统计学知识。为了使用户能够轻松快速地进行代谢组学数据分析,我们为代谢组学工作流程提供了全面的解决方案(图 S1)。
标准代谢物分析工作流程包括5个步骤:(1)数据预处理:方法主要包括对原始数据缺失值的过滤、缺失值估计、数据归一化、质量控制(QC)验证和数据转换。(2)样本比较分析:采用PCA和PLS-DA进行多因素统计分析;(3)代谢物注释:代谢物在KEGG(http://www.genome.jp/kegg/)和HMDB(https://hmdb.ca/)数据库中注释;(4)差异表达代谢物分析:采用多维分析与单维分析相结合的方式筛选组间差异代谢物;(5)代谢集分析:关键或差异表达代谢物的分析和可视化,如代谢物聚类、相关性分析等。此外,我们还提供了一些高级分析来揭示生物过程的奥秘,例如:随机森林、支持向量机等。
多组学工作流
多组学技术有助于研究人员揭示疾病病理生理学以及临床表型的潜在机制。多组学提供了跨多个层面的综合视角,而单一组学数据只能部分解释复杂生物过程的单一层面。转录组学和蛋白质组学数据关联分析流程支持差异表达分析、mRNA与蛋白质丰度之间的相关性、功能注释和富集、GSVA,以及交互式可视化,包括维恩图、象限图、九象限图、气泡图、箱形图和甜甜圈图。该流程可实现综合互补的研究视角,从而全面了解从mRNA到蛋白质的生物分子过程。
微生物组和代谢组关联分析工作流程可用于分析物种/功能与代谢物之间的关联,从而帮助建立“物种/功能-代谢物-表型/靶器官”之间的逻辑关联。研究结果系统地描绘了不同维度的生物过程的调控机制。为了便于直观地呈现科学发现,工作流程提供了丰富的分析内容。主要分析内容如下:(1)查阅单组学特征集的注释信息和丰度信息(物种、KO基因和代谢物种);(2)通过Procrustes和O2PLS分析微生物群落与代谢物之间的协同作用,筛选出对区分不同组样品贡献最大的物种和代谢物。(3)为了解释关键菌群与代谢产物的关联性,可通过HCLUST分析、Mantel Test网络热图、表达相关热图和弦图、表达相关网络、线性回归分析、MaAsLin分析和典型相关分析(CCA)来实现。(4)利用微生物组和代谢组数据形成组合标志物,通过随机森林、支持向量机(SVM)、LASSO和logistic回归等4种集成机器学习算法,高效筛选预测性生物标志物。此外,MIMOSA2、mmvec和WGCNA可用于进一步分析,以解释微生物和代谢物之间可能的相互作用。(5)代谢物检测技术用于检测中间代谢物,结合宏基因组数据预测的代谢途径,可以重构下游代谢途径,以获得完整的微生物代谢途径。
“工作流+拓展工具”,一种新的交互式分析模式
为了改善用户体验并拓展分析深度,我们开发了一种全新的交互式分析模式。以真核生物参考转录组分析流程为例,用户可以选择工作流中生成的数据表,并在扩展工具中设置参数,完成更深入的数据挖掘。将工作流生成的中间数据提取为JSON格式的参数,通过base64加密传输到特定工具,并在分析工具中完成参数解析(图S2)。真核参考转录组分析流中提供了28款拓展工具可供用户使用,包括IGV可视化、差异表达基因雷达图、双曲线火山图、Circos图、单基因GSEA、多通路GSEA等。
用户和论文发表情况
自2016年10月美吉生物云发布以来,来自9,000+所知名高校和机构的150,000+科研用户,在云上完成了超过600,000项组学数据挖掘任务。2024 年,2,015篇期刊文章在方法学中提及了 Majorbio Cloud。目前,单细胞转录组学工作流、蛋白质组学工作流和代谢组学工作流分别助力科研工作者发表了20, 62, 393篇学术论文。我们将不断更新和迭代平台,服务我们的用户更深入地开展组学数据分析研究。
作者简介

韩继臣(第一/通讯作者)
● 就职于上海美吉生物医药科技有限公司,齐齐哈尔大学遗传学硕士。
● 2009年起从事组学技术在生命科学研究中的应用推广工作,领导主责开发及推广的美吉生物云平台,为超过9,000+国内外科研单位和企业,150,000+科研工作者提供一站式组学生信分析服务。

韩畅(第一/通讯作者)
● 哈尔滨工业大学生物学硕士,就职于上海美吉生物医药科技有限公司。
● 负责蛋白组学和转录组学的数据分析和挖掘与云平台产品的研发设计。发表学术论文9篇,相关成果取得国家发明专利1项,软件著作权2项。承担和参与上海市经信委、上海市科委课题3项。

史彩萍(第一作者)
● 华东师范大学生物信息学硕士,就职于上海美吉生物医药科技有限公司。
● 主要负责转录组学、单细胞组学、蛋白组学以及多组学联合等云平台产品分析流程以及云工具的研发与开发,拥有10多年上述组学数据分析经验,参与发表SCI文章6篇,专利5篇,获批软著8项。

刘林梦(第一作者)
● 上海交通大学生物学硕士,就职于上海美吉生物医药科技有限公司。
● 目前研究方向是微生物组、微生物比较基因组以及多组学联合的数据分析与挖掘,数据库和软件研发经验丰富。部分成果发表于Nucleic Acids Research、The ISME Journal、iMeta等期刊。
更多推荐
(▼ 点击跳转)
iMeta | 引用7000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期

3卷1期

2卷2期封底

2卷4期封底

3卷2期

3卷3期

3卷3期封底
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF 23.7)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、50万用户的社交媒体宣传等。2022年2月正式创刊发行!目前期刊已经被ESCI、PubMed、Scopus等数据库收录。
联系我们
iMeta主页:
http://www.imeta.science
姊妹刊iMetaOmics主页:
http://www.imeta.science/imetaomics/
出版社iMeta主页:
https://onlinelibrary.wiley.com/journal/2770596x
出版社iMetaOmics主页:
https://onlinelibrary.wiley.com/journal/29969514
iMeta投稿:
https://wiley.atyponrex.com/journal/IMT2
iMetaOmics投稿:
https://wiley.atyponrex.com/journal/IMO2
邮箱:
office@imeta.science

















 969
969

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









