






点击蓝字 关注我们
16S rRNA基因扩增子测序技术的漫长发展旅程正迈向基于个体的菌群功能解析

综 述
● 原文链接DOI: https://doi.org/10.1002/imo2.9
●2024年7月11日,中国科学院动物研究所金坚石团队在iMetaOmics在线发表了题为“Long journey of 16S rRNA-amplicon sequencing toward cell-based functional bacterial microbiota characterization”的文章。
● 本综述梳理了16S rRNA基因扩增子测序方法的重要发展历程,总结了当前16S rRNA基因扩增子测序技术的特性和应用范围,还展望了这一领域未来技术发展的方向。
● 第一作者:金坚石
● 通讯作者:金坚石(jinjs@ioz.ac.cn)、城口克之(katsuyuki.shiroguchi@riken.jp)
● 合作作者:刘雄舵
● 主要单位:中国科学院动物研究所、日本理化研究所BDR中心(RIKEN Center for Biosystems Dynamics Research)
亮 点
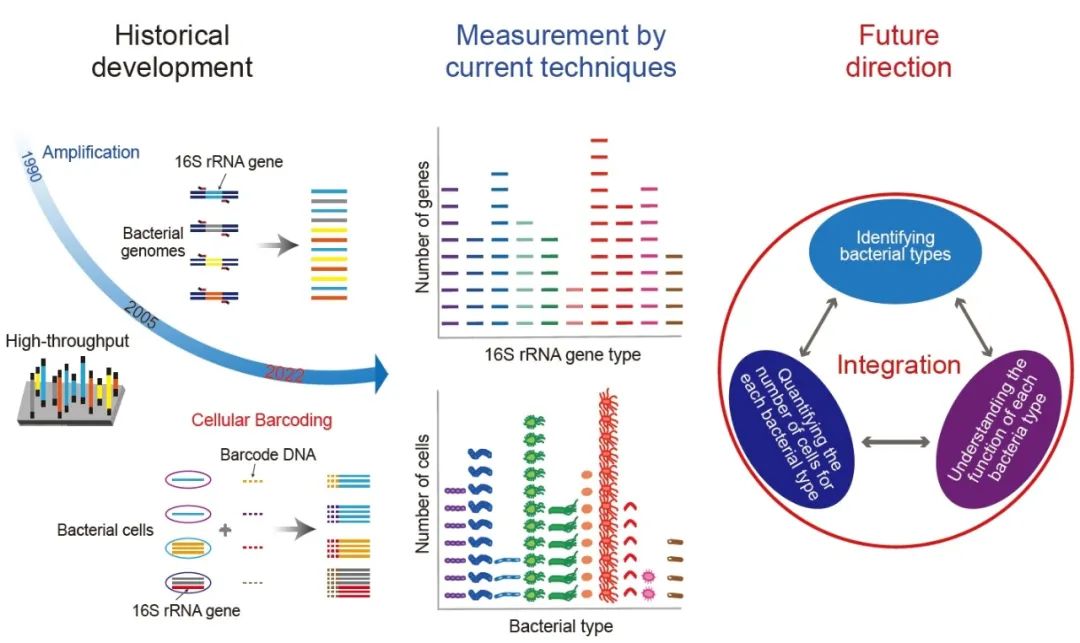
● 总结了16S rRNA基因扩增子测序技术的发展历史;
● 比较了目前不同16S rRNA基因扩增子测序技术的特性;
● 最新技术使得细菌个体的鉴定和每种细菌数量的定量分析成为可能;
● 展望了这一领域未来发展的重点是如何解析细菌个体在菌群中的功能。
摘 要
细菌通常以群落形式存在并行使功能,该群落被称为细菌菌群,通常含有大量来自不同物种的细菌。因此对细菌菌群的解析需要能高通量鉴定与定量大量细菌个体的方法,为建立这一方法,领域已努力开发了30多年。这篇综述首先介绍了可高通量解析细菌菌群的16S rRNA基因扩增子测序技术的发展历史。接着总结了当前16S rRNA基因扩增子测序技术的特性和应用范围,包括最近开发成功的具有单碱基精度并能高通量鉴定和定量细菌个体的新技术。在此基础上,该综述还展望了这一领域未来技术发展的方向,包括联合使用高通量定量方法和其他如具有细菌个体区分度的全基因组测序技术等其他能获得更丰富信息的分析方法,实现对细菌菌群包括鉴定、定量和功能分析的全面解析。
全文解读
引 言
在海洋或陆地环境中,细菌通常以菌群的形式与其他生物(如人、动物、植物)共生。近年来,人们对操纵环境细菌菌群以恢复生态系统功能或操纵共生细菌以恢复宿主健康的兴趣越来越大。然而,要理解细菌菌群与环境或生物体的相互作用或者对细菌菌群进行操纵,首先要解决的问题是解析细菌菌群的组成,即细菌菌群中存在多少种细菌和每种细菌的数量是多少。
由于基因组记录了进化信息,通过基因组序列的比对分析可以有效地分类细菌。然而,即使使用最先进的技术,对菌群中所有细菌进行全基因组测序也是不可行的。因此,在一次实验中,需要在每个细菌个体测定多长基因组和检测多少细胞数之间进行权衡。尽管通过全基因组测序或宏基因组测序可以提供基于基因的功能信息,但在对包含大量细菌的菌群进行高通量分析时,只对每个细菌的单个基因或单个基因的部分进行分析是最高效的。自1977年起,16S rRNA基因(约1500个碱基对)因其以下特征被广泛应用于细菌的系统分类:1)存在于所有细菌中;2)包含9个可变区(V1-V9)可以有效地区分细菌种类;3)在可变区之间存在高度保守区域可作为纯化或扩增16S rRNA基因的靶点。为了利用16S rRNA基因解析细菌菌群的组成,自1985年以来,人们一直在开发可用于全面解析菌群中16S rRNA基因的测序方法。以下是大多数已发明的16S rRNA基因扩增子测序方法的标准流程:通过一对靶向16S rRNA基因保守区域的通用引物扩增菌群中细菌的16S rRNA基因,并利用高通量测序平台测序获得所有扩增的16S rRNA基因的序列。
高通量的16S rRNA基因扩增子测序方法有以下优点:该方法基于16S rRNA基因的扩增,因此可以适用于未培养的细菌或低生物量样品,并且可以对复杂样品进行细菌特异性的分析。因此,该方法在医学、农学、环境科学等多个领域的研究中都有广泛的应用。在医学领域,16S rRNA基因扩增子测序方法可用于鉴定口咽或直肠中特定的病原菌,用于流行病或传染病分析;还可用于检测低生物量样品(如血液或肿瘤环境)中的特定细菌;同时,它还可用于脑脊液和呼吸道样本中细菌的鉴定,为抗生素使用计划提供参考。在农学领域,16S rRNA基因扩增子测序方法可用于新鲜农副产品上的病原菌检测,以确保食品安全,并可用于细菌与植物的相互作用研究。在环境科学研究中,16S rRNA基因扩增子测序方法可用于分析废水、湖水等环境样品的细菌菌群组成,以评估环境污染和生态修复效果。
本综述从梳理16S rRNA基因扩增子测序方法的重要发展历程开始,对目前用于鉴定与定量16S rRNA基因或细菌类型的16S rRNA基因扩增子测序方法的特性进行了比较和总结。接着,概述了16S rRNA基因扩增子测序方法的最新研发进展与应用拓展。最后,本综述还对16S rRNA基因扩增子测序方法的未来发展方向进行了讨论。
16S rRNA基因扩增子测序方法研发历程的里程碑
首先,16S rRNA基因扩增子测序实验方法的研发有以下几个重要的时间节点(图1)。1985年,Pace等人首次通过测序16S rRNA基因序列来分析细菌菌群的组成。他们针对16S rRNA基因的可变区之间的保守区域设计了DNA探针并通过探针杂交进行16S rRNA基因的纯化富集,然后通过DNA克隆扩增和 Sanger测序进行测序分析;中间无需培养细菌。1990年,更高效的16S rRNA基因PCR扩增富集法取代了DNA探针杂交富集法,即使用针对16S rRNA基因保守区域设计的引物特异性PCR扩增16S rRNA基因。这一富集方法仍然是当前16S rRNA基因扩增子测序方法的关键步骤。2006年,下一代测序技术(二代测序)首次应用于16S rRNA基因扩增子测序。虽然下一代测序技术只能检测最多600 bp左右长的16S rRNA基因序列,但它对能够检测的基因数量提高了几个数量级,由于细菌菌群中存在大量细菌,因此通量的提升对于细菌菌群的分析至关重要。此外,使用下一代测序技术后无需再做DNA克隆,简化了16S rRNA基因扩增子测序的步骤。因此目前主流的16S rRNA基因扩增子测序方法仍采用下一代测序技术。2013年,16S rRNA基因扩增子测序技术采用单分子测序技术(三代测序)实现了对16S rRNA基因全长序列的高通量测序。2022年,基于单细胞的16S rRNA基因扩增子测序方法研发成功,被称为BarBIQ(Barcoding Bacteria for Identification and Quantification)法。该方法通过对每个细菌细胞的16S rRNA基因进行条形码标记和测序,实现了对细菌菌群中细菌细胞类型的鉴定和细菌个数的定量。
除了实验技术的发展,还有一系列用于纠正16S rRNA基因扩增子测序方法中由于扩增和测序产生错误的分析方法被陆续开发出来。1995年,首次出现了通过序列聚类的方法来纠正测序与扩增错误的技术手段。随后,许多针对测试数据的聚类算法被提出,直到现在仍被广泛使用。然而,基于序列聚类的方法存在一个问题,即聚类的结果会依赖于整个数据集,因此很难直接进行不同研究之间的比较。2013年,一种通过统计分析直接从测序数据中识别错误序列和正确序列的算法被提出,不再需要对序列的聚类,实现了不同数据集之间的直接比较。此外,为了整合不同的分析方法实现对16S rRNA基因扩增子测序数据的全面分析,综合分析平台mothur与QIIME分别于2009年和2010年建立并一直更新,至今仍被广泛使用,这些平台的建立也促成了这一领域数据格式的全球标准化。
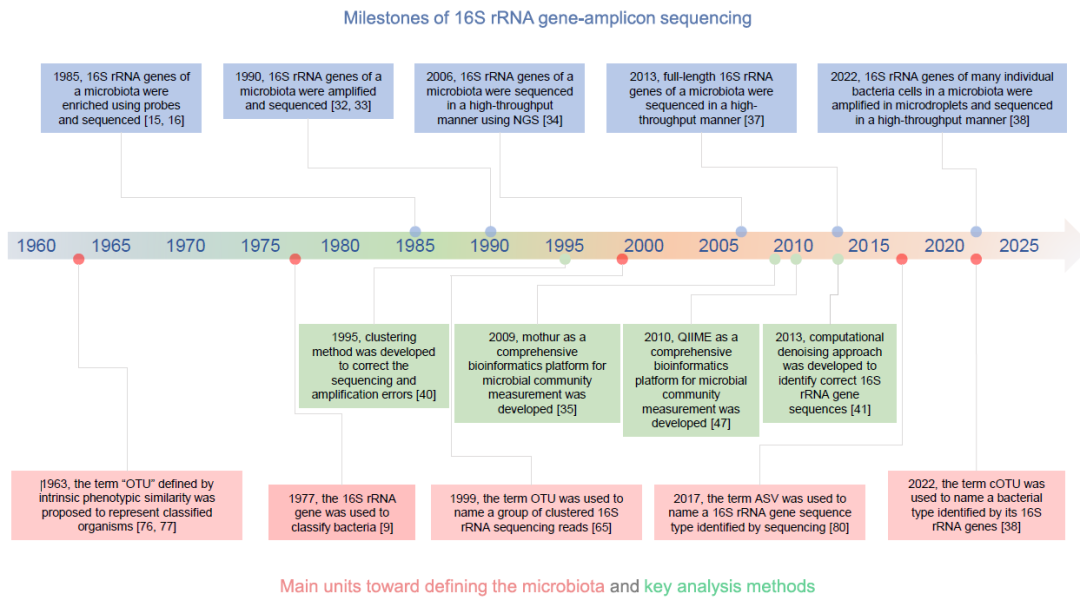
图1. 16S rRNA基因扩增子测序技术的历史
16S rRNA基因的鉴定
鉴定菌群中的细菌类型是16S rRNA基因扩增子测序方法的主要目标。然而大多数方法目前只能鉴定菌群中的16S rRNA基因,而一个细菌细胞可能含有多种不同的16S rRNA基因,因此这类方法的结果并不能完全反映菌群中的细菌类型。这部分,我们将介绍几种当前还在使用的鉴定16S rRNA基因序列的方法。
当前用于鉴定16S rRNA基因的方法
目前广泛使用的16S rRNA基因扩增子测序方法都是基于下一代测序和单分子测序技术,以低成本进行高通量的分析。然而,由于这些方法存在较大的测序和扩增错误,使得正确识别16S rRNA基因序列变得困难。为了纠正测序与扩增错误,近几十年来开发了一系列方法,包括序列聚类法、统计分析去噪以及分子条形码标记法。
首先是序列聚类法。在过去几十年广泛使用的16S rRNA基因扩增子测序方法中,菌群中的16S rRNA基因被扩增并测序。接着根据序列相似度和设定的阈值,采用不同的聚类算法将测序得到的序列聚类成不同的组。最后,选择丰度最高的16S rRNA基因序列来代表同一聚类组中的所有序列。这种序列聚类法基于的理论假设是相同分类细菌具有相似的16S rRNA基因序列,并且测序以及扩增出现的序列错误概率较低。因为序列聚类法需要对序列进行聚类,所以无法区分序列相近的16S rRNA基因。
据我们所知,序列聚类法于1995年被首次使用,并从1999年后被广泛沿用至今。一开始,不同文献采用不同的术语来描述序列聚类得到的聚类组。在2001年的一篇综述中,统一选择了“OTU(operational taxonomic unit)”这个术语来描述不同文献通过序列聚类得到的聚类组,即分类单元。后来,越来越多的分析软件也开始使用了OTU这个术语,比如DOTUR、USEARCH、mothur、QIIME以及EasyAmplicon等。现在, OTU已成为16S rRNA基因测序数据序列聚类分析方法的全球标准术语。
关于OTU序列聚类法已经被多次综述和总结,主要有三种:de novo OTU法、closed-reference OTU法和open-reference OTU法。在closed-reference和open-reference OTU法中,数据库被用于序列聚类的核心序列以控制序列纠错的准确性,因此数据库的开发与更新也在不断进行,以提高对OTU鉴定的准确性。比如,Greengenes2, SILVA (更新至2020) , MiDAS 4, MetaSquare, RiboGrove, forensic microbiome database数据库和comprehensive ecosystem-specific reference databases数据库在近几年被开发出来,有些主要针对某个特定领域,另一些则整合了16S rRNA基因序列数据和细菌全基因组数据。
OTU这个术语最早在1963年被用来描述基于内在表型相似性来分类的生物类群,这与16S rRNA基因扩增子测序方法中的定义不同,后者指基于序列相似性聚类产生的一组基因序列。然而,由于某些细菌的基因组中存在多个16S rRNA基因序列,并且这些序列可能存在显著差异(比如有报道称序列相似性可能小于95%),因此特定细菌的16S rRNA基因可能会被聚类在不同的OTU中。这意味着在16S rRNA基因扩增子测序方法中,鉴定的OTU代表的是聚类得到的基因序列,而不是原先‘OTU’所定义的生物个体类群。因此,OTU聚类法并不能通过16S rRNA基因分类细菌生物个体。
统计分析去噪法。去噪法是为了纠正测序和扩增错误(包括嵌合体)而开发的一种统计分析算法,其中包括DADA2等多种去噪算法。最初,去噪法被应用于下一代测序技术产生的测序数据,后来也被应用于单分子长读长测序获得的数据。去噪法基于测序得到的序列中错误序列相对正确序列概率低很多的假设,通过统计分析找到错误序列,从而识别正确的16S rRNA基因序列,通过这些方法获得的序列类型被称为ASV(amplicon sequence variants)。虽然去噪方法的纠错策略很有效,但仍不是完美的。例如,在只有9-20株细菌的低复杂菌群中,DADA2并不能完全纠正所有的错误。需要说明的是,去噪法也是用来鉴定16S rRNA基因类型的方法,在这一点上和最早出现在1995年的聚类法是一致的。但是,用基于ASV或OTU分析方法对同一数据进行分析,通常会得到明显不同的16S rRNA基因序列结果。
最后是分子条形码标记法。为了提高序列鉴定的准确度,DNA分子条形码在2013年被首次应用于16S rRNA基因扩增子测序方法。这种方法将每个16S rRNA基因连接到一个独特的分子条形码(即不同的DNA序列)上,然后扩增连接了分子条形码的序列。因此,基于含有错误的序列少于正确的序列的假设,在测序之后,可以通过比较连接了相同分子条形码的16S rRNA基因测序序列(来自同一16S rRNA基因)来纠正测序错误。虽然分子条形码可以纠正测序错误,但是无法解决16S rRNA基因扩增子测序方法面临的另一个主要问题,即扩增错误。
16S rRNA基因鉴定方法的最新进展拓展了其应用范围
近年来, 16S rRNA基因扩增子测序方法在DNA提取、引物设计、DNA扩增和生物信息学工具等方面得到了改进,扩大了其应用范围。首先,针对低细菌生物量的样品如血液、组织等开发了针对性的DNA提取方法。16S rRNA基因DNA富集方法也被用于临床标本和肿瘤组织等极低细菌生物量的样品。
其次,针对不同的16S rRNA基因可变区建立了不同的引物组合,通过对不同引物扩增结果的比较,提出对于不同的研究需要选择不同的靶向区域以得到更好的结果。此外,为了得到更高的分辨率,建立了针对全长16S rRNA基因以及相邻基因的引物。除了通用的引物外,还有很多针对特异细菌群的引物被设计出来,可以应用于特殊样品的检测,比如北极菌群、呼吸菌群、马肠道菌群、淡水菌群和海沟沉积物菌群等。重要的是,这些技术的进步促成了很多新细菌类群的发现,比如发现了阿斯加德古细菌、蛭弧菌和布罗卡迪亚厌氧氨氧化菌等。
第三,改进了测序文库的扩增方法。在基于下一代测序技术的方法中,如何得到高效特异的扩增结果也有很多研究。数字PCR被应用于增加低生物量样本的扩增效率。这种方法在16S rRNA基因扩增子测序分析中实现了对于低DNA浓度样本的准确分析。此外,长读长测序技术LoopSeq被应用于16S rRNA基因扩增子测序中,可以准确鉴定全长16S rRNA基因序列。使用该方法,在零售肉类样品中的同一菌种中鉴定出了不同的菌株,确定了潜在的病原体。
第四,为了提高全长16S rRNA基因序列鉴定的准确性,一系列基于数学算法的生物分析软件如MetaMaps、Emu、NanoCLUST以及16S-FASAS被开发出来。改进的16S rRNA基因扩增子测序方法Emu成功得到了与全基因组鸟枪宏基因组方法相同的结果,即将12个阴道拭子的细菌群落分类为6种“群落状态类型”。
基于16S rRNA基因的细胞类型鉴定
对菌群中细菌个体的分类鉴定是理解菌群功能的基础。目前常用的鉴定方法是通过培养组学培养出单细菌之后对其基因组或16S rRNA基因进行测序。这种方法高度依赖对细菌的分离培养,然而,目前大量的细菌仍无法被培养。此外,这种方法的通量较低,很难进行菌群水平的高通量解析。
正如我们上面讨论的,不依赖培养的16S rRNA基因扩增子测序方法是一种高通量方法。然而,由于许多细菌的基因组中含有多种不同的16S rRNA基因序列,通过传统方法鉴定的16S rRNA基因类型通常不能代表细菌类型。
为了对菌群中的细菌类型进行鉴定,基于细胞的16S rRNA基因扩增子测序方法(BarBIQ)最近被成功开发。BarBIQ可以高通量地测定单个细菌中的16S rRNA基因序列,并且拥有单碱基精度以及分辨率;一次实验可测定超过105 个细菌。BarBIQ定义的分类单元是cell-based operational taxonomic unit(cOTU)。cOTU与OTU以及ASV有本质的不同(图2)。对于单细胞生物,cOTU与最开始的OTU(一类表型类似的生物体)的概念相同。
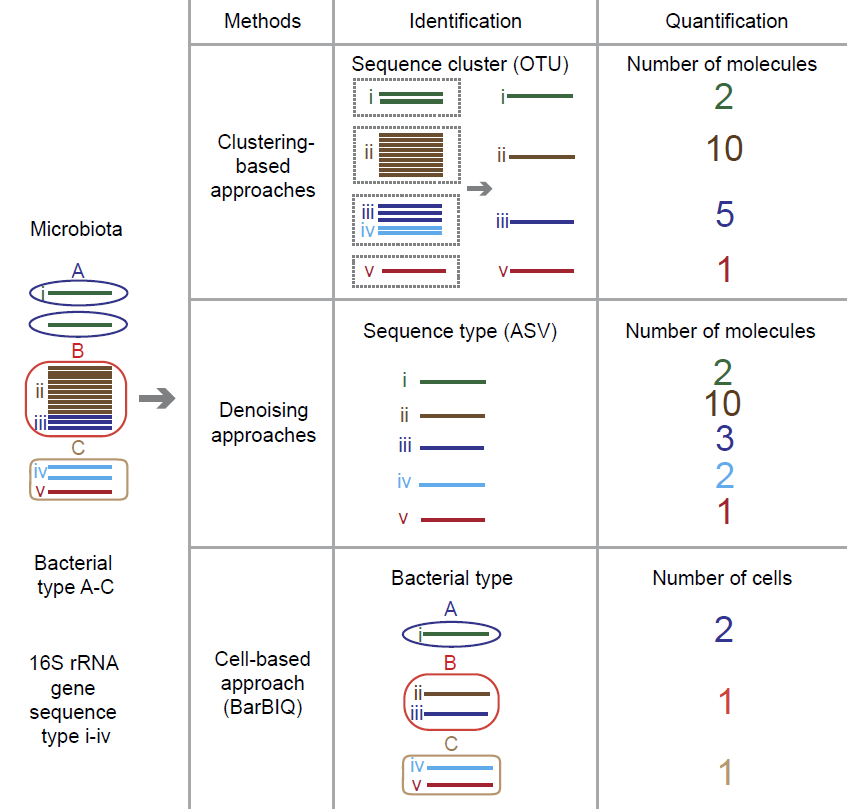
图2. 16S rRNA基因扩增子测序方法的比较
16S rRNA基因的定量
对每种细菌的定量是16S rRNA基因扩增子测序方法的另一个目标。目前大部分方法定量的是16S rRNA基因的数量,然而由于不同的细菌有不同的16S rRNA基因拷贝数(1-15),因此这些方法定量的并不是细菌细胞(个体)的数量。这部分将讨论定量16S rRNA基因数量的方法。
扩增的16S rRNA基因DNA分子(测序读段)的定量
16S rRNA基因扩增子测序是把每个16S rRNA基因扩增为多个DNA分子后测序,即测序得到的每条读段(read)代表一个扩增后的DNA分子。因此,通过统计被聚类到每个OTU或归类到每个ASV中的读段数即可统计出被测定的每个被鉴定的OTU或ASV的DNA分子数量。如果用每个检测样品中检测到的总DNA分子数进行归一化,即可得到每个OTU或ASV的相对丰度。然而,由于不同的DNA序列(不同的16S rRNA基因序列)被扩增的效率不同,每种16S rRNA基因序列被扩增出的DNA分子数量对的相对丰度与扩增前的16S rRNA基因数量的相对丰度并不一致。此外,对同一数据使用不同的分析工具分析也可能得到不同的结果,即16S rRNA基因DNA分子的相对丰度会有差异。
扩增前的16S rRNA基因的定量
了消除16S rRNA基因定量中扩增引起的偏差对结果的影响,分子条形码标记技术被引入16S rRNA基因扩增子测序技术。该技术通过对每个16S rRNA基因连接上不同的分子条形码来标记DNA分子扩增自哪个16S rRNA基因,因此通过对条形码类型的计数即可得到扩增前的16S rRNA基因的数量。
通过分子条形码标记并结合理论预测算法已经可以确定样品中16S rRNA基因的绝对数量。在这种预测方法中,作者假设PCR扩增是一个随机过程,并通过泊松分布计算,根据每种基因类型的16S rRNA基因的测量数量来估计样品中16S rRNA基因的绝对数量。
近些年来,一些研究者还尝试通过使用内部参照、总分子浓度或偏差估计来归一化OTU相对丰度的途径来获得OTU的绝对丰度。然而,这些方法尚未与分子条形码标记法相结合。因为16S rRNA基因分子的相对丰度是由扩增后的DNA分子数的相对数量决定的,而扩增的DNA分子数会受到扩增偏差与噪音的影响,因此所测得的结果并不能准确反映扩增前16S rRNA基因的数量,这使得准确定量样品中16S rRNA基因的绝对数量变得困难。
细菌的绝对定量
定量菌群中的细菌数量是理解菌群的基础。由于每个细菌含有多条16S rRNA基因拷贝且不同细菌拷贝数不同,通过对16S rRNA基因的定量很难确定细菌的数量。有一些研究试图通过生物信息学的方法解决这一问题。这种方法的主要思路是利用每种细菌含有16S rRNA基因的拷贝数来换算测定的16S rRNA基因数以获得细菌数量。然而许多细菌基因组中的16S rRNA基因拷贝数是未知的,因此很多软件通过假设相同类型的细菌物种含有相同16S rRNA基因拷贝数来把未知细菌的基因数换算成细菌数。但是,很多情况下,同一类细菌中不同物种细菌的16S rRNA基因拷贝数也不一定相同,因此很难用生物信息学方法从未知细菌的16S rRNA基因数换算出该细菌的数量。
为了实验测定菌群中细菌的数量,基于单细胞的BarBIQ方法近期被开发出来。BarBIQ对每个细菌细胞的16S rRNA基因标记上DNA条形码,因此对条形码类型数量的统计即可计算出每种细菌类型的相对个数。接着,通过数字PCR测定每单位重量或体积中的总细菌数,并对测定的相对细菌个数归一化,即可获得单位重量或体积样品中被鉴定的每种细菌类型(cOTU)的绝对细菌数。
一些研究也尝试使用流式细胞仪或使用已知数量的内参对细菌总数量进行定量,然后通过对OTU的相对丰度进行归一化来实现对细菌个体数量的绝对定量。在这些研究中,细菌的相对数量是通过每种细菌中16S rRNA基因的拷贝数来换算OTU的相对数量获得的。然而,正如之前提到的,OTU并不能代表细菌类型,同时目前对未知细菌中的16S rRNA基因的拷贝数还无法准确估计,因此使用以上基于OTU的方法并不能实现细菌个体数的绝对定量。
结论与展望
目前的16S rRNA基因扩增子测序方法已经能够实现对细菌类型的鉴定与定量,以及全长16S rRNA基因的检测。然而16S rRNA基因扩增子测序方法仍存在一些不足。首先,由于16S rRNA基因扩增子测序方法只对一个基因进行检测,因此很难实现高分辨率的鉴定,例如菌株水平的鉴定。这个问题可以通过多基因分析来解决。其次,在样品收集与储存过程中使用不同的条件,如不同的温度和时间等,可能会导致细菌生长或死亡,从而改变样品中的细菌组成。因此,开发针对不同样品的标准化的采样和储存方法至关重要。第三,对于细菌生物量低且宿主DNA量高的样品来说,减少宿主基因污染是主要难点。尽管目前已经有一些有效的方法,但对不同的样品需要定制化设计和优化。因此,针对低细菌生物量的样品后续需要开发通用且简单的样品处理方法。
理想情况下,对菌群进行全面的解析策略是对菌群中所有细菌进行单细菌全基因组测序,以同时获得菌株水平的细菌组成、数量以及功能信息。然而,由于测序仪通量和成本的限制,很难通过这种方式解析菌群。因此,出现了以下假设:如果鉴定到的细菌16S rRNA基因序列与数据库中全基因组测序得到的16S rRNA基因序列能匹配上,那么菌群中的这些细菌的全基因组就能被获得。然而,由于细菌全基因组的数据库仍不完善,上述假设可行性不高。一种可能有效的策略是将BarBIQ与单细胞全基因组测序相结合:通过BarBIQ在单细菌水平鉴定出某个具有目标特性的细菌,然后基于该细菌的16S RNA基因靶向分选该细菌,并进行全基因组测序。如果是随机挑选菌群中的细菌进行单细胞全基因组测序,对于丰度低的细菌可能会很难获得足够的数据,而如果是按上述设想针对性地选择细菌进行测序,那将不会受到菌群中细菌丰度的限制。
此外,利用最近报道的新方法,通过高通量筛选出的细菌可能可以分离出有活性的细菌。如果可以分离出活细菌,则很多功能分析技术都可被用来研究靶向细菌,如代谢物检测技术和单细胞转录组分析技术。当然实现这一目标,后续可能需要进一步提升这些技术的检测灵敏度或者扩增被分离的细菌数量。
一旦上面提到的检测可以实现,基于细菌个体数量以及细菌功能进行数学建模将有助于理解与预测菌群的行为。这将为医学和农业等多个领域提供有效的工具,同时也可以预测粪便移植或者施加农药等操作下菌群的响应。
总之,最近研发成功的基于高通量16S rRNA基因扩增子测序的准确鉴定和定量菌群中细菌个体的方法,提供了一个可以整合鉴定、定量与上面提到的功能分析的途径,将开启一个深入理解菌群功能的新时代(图3)。
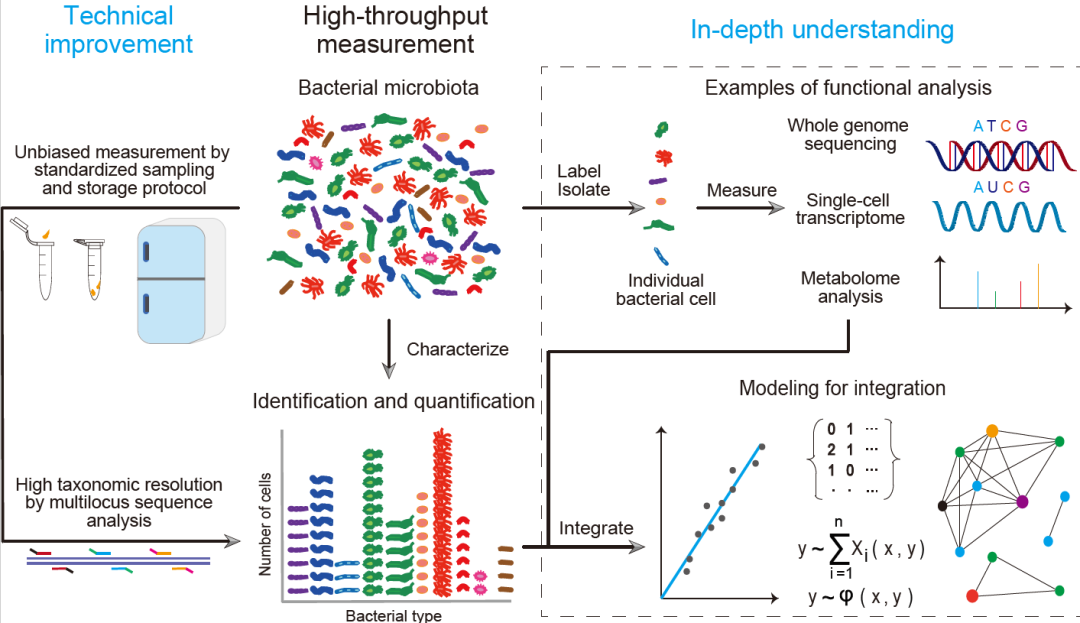
图3. 更深入了解细菌菌群的局限与展望
引文格式:
Jianshi Jin, Xiongduo Liu, Katsuyuki Shiroguchi. Long journey of 16S rRNA-amplicon sequencing toward cell-based functional bacterial microbiota characterization. iMetaOmics 9: e9 https://doi.org/10.1002/imo2.9
作者简介

金坚石(第一作者)
● 研究员,博士生导师,中国科学院动物研究所农业虫害鼠害综合治理研究国家重点实验室“菌群生物学与智能调控研究组”组长。
● 2009年和2014年分别获南京大学学士和北京大学博士学位,2010-2011哈佛大学访问学者,2014-2023年在北京大学和日本理化学研究所先后担任博士后、JSPS访问研究员、Senior Scientist,2023年3月全职加入中国科学院动物研究所。课题组从事面向解决重要菌群生物学问题的精准、高通量、智能化系统研发工作,聚焦哺乳动物的共生菌群功能研究。研究成果发表于Science,Nature Communications, Nature Protocols,Science Immunology,PNAS等杂志。已申请多国PCT国际专利。曾获得日本理化学研究所“荣峰奖”、“樱舞奖”、日本JSPS政府奖学金、北京大学优秀博士论文,北大-清华生命科学联合中心优秀博士后基金等。
● 金坚石课题组长期招聘博士后与助理研究员、副研究员和工程师(均为中科院编制)。欢迎具有生物实验技术、生物信息学、微生物学、动物学、免疫学、分子生物学、生物物理学或计算机等相关专业的博士或硕士申请。同时非常欢迎联合培养的研究生来课题组进行合作研究,有意向报考研究生和博士生的同学来研究组深入了解。
iMetaOmics
更多资讯
● iMeta姊妹刊iMetaOmics(定位IF>10)欢迎投稿!(2024.2.27)
● iMeta姊妹刊iMetaOmics编委招募 (定位IF>10) (2024.3.2)
● iMeta姊妹刊iMetaOmics电子版和印刷版ISSN申请获批(2024.4.1)
● iMeta姊妹刊iMetaOmics投稿系统正式上线(2024.4.17)
● iMeta姊妹刊iMetaOmics主编正式官宣(2024.4.22)
● 出版社iMetaOmics主页正式上线!(2024.4.28)
● iMetaOmics | 浙江大学宗鑫组揭示两猪种宿主-肠道菌群互作差异
● iMetaOmics | 罗鹏/袁硕峰/苗凯/程全发表STAGER: 生成式人工智能可靠性的标准化测试和评估推荐
● iMetaOmics | 徐州医科大杨欢组揭秘沙门氏菌-宿主-微生物群在免疫与代谢中的相互作用
更多推荐
(▼ 点击跳转)
iMeta | 引用7000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期

3卷1期

2卷2期封底

2卷4期封底

3卷2期

3卷3期

3卷3期封底
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百千华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 20)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!发行后相继被Google Scholar、ESCI、PubMed、DOAJ、Scopus等数据库收录!2024年6月获得首个影响因子23.7,位列全球SCI期刊前千分之五(107/21848),微生物学科2/161,仅低于Nature Reviews,同学科研究类期刊全球第一,中国大陆11/514!
“iMetaOmics” 是“iMeta” 子刊,主编由中国科学院北京生命科学研究院赵方庆研究员和香港中文大学于君教授担任,定位IF>10的高水平综合期刊,欢迎投稿!
联系我们
iMeta主页:
http://www.imeta.science
姊妹刊iMetaOmics主页:
http://www.imeta.science/imetaomics/
出版社iMeta主页:
https://onlinelibrary.wiley.com/journal/2770596x
出版社iMetaOmics主页:
https://onlinelibrary.wiley.com/journal/29969514
iMeta投稿:
https://wiley.atyponrex.com/journal/IMT2
iMetaOmics投稿:
https://wiley.atyponrex.com/journal/IMO2
邮箱:
office@imeta.science

















 2万+
2万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









