






点击蓝字 关注我们
高效易用GutHi-C技术:捕获菌群宏基因组三维(3D)互作

研究论文
● 期刊: iMeta (IF 23.7)
● 原文链接DOI: https://doi.org/10.1002/imt2.227
● 2024年7月22日,中国农业科学院孔思远和潘玮华团队在iMeta在线联合发表了题为“Efficient and easy-to-use capturing three-dimensional metagenome interactions with GutHi-C”的文章。
● 本文开发了GutHi-C技术,该方法高效、快速,可广泛应用于人类、家畜和家禽肠道微生物,开拓了微生物宏基因组Hi-C重要技术手段。
● 第一作者:卢宇曦、杨金宝、李晨莹
● 通讯作者:孔思远(kongsiyuan@caas.cn)、潘玮华(panweihua@caas.cn)、朱秀生(zhuxiusheng@caas.cn)
● 合作作者:田蕴涵、常荣荣、孔大帅、杨述林、王彦芳、张玉波
● 主要单位:中国农业科学院深圳农业基因组研究所(岭南现代农业科学与技术广东省实验室深圳分中心)、河南大学生命科学学院、华中农业大学信息学院、青岛农业大学动物科学与技术学院、中国农业科学院北京畜牧兽医研究所、美国弗雷德里克癌症研究国家实验室
亮 点
Bilibili:https://www.bilibili.com/video/BV1rDs5eaEsx/
Youtube:https://youtu.be/aYEhhRO3eBk
中/英文操作视频链接
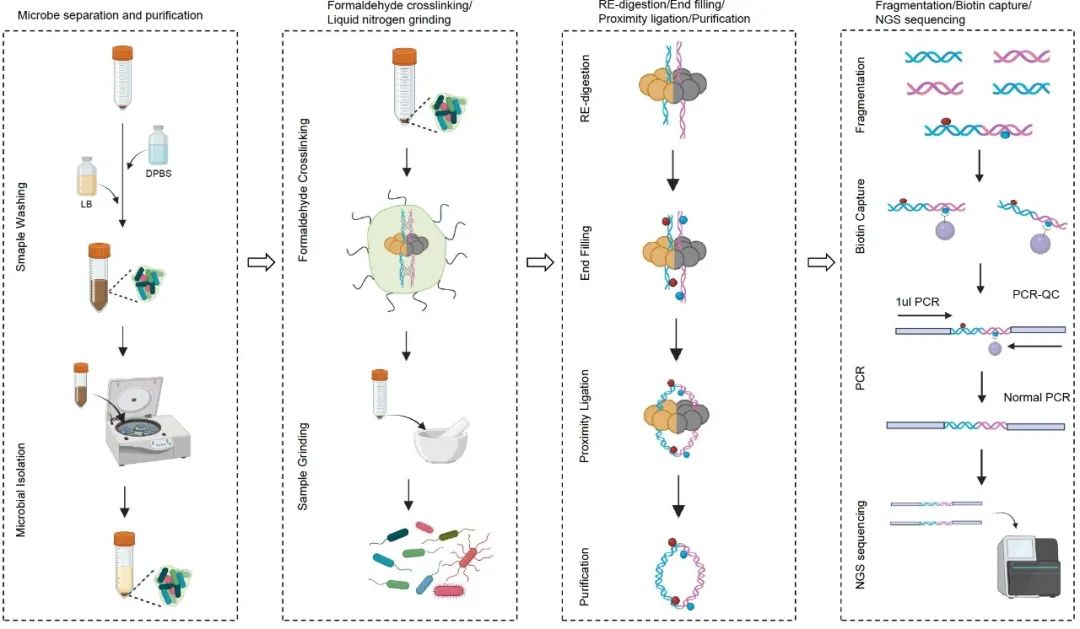
● 提供了一种高效、易于操作的GutHi-C 宏基因组三维互作捕获技术策略,适用于肠道微生物组等微生物群体;
● 介绍了GutHi-C 技术的操作流程,该方法节约了试剂成本,提高了文库构建的成功率、质量以及测序数据质量;
● GutHi-C 可以应用于揭示微生物群体单菌水平的三维构象,还可以辅助高保真的成环的完整宏基因组组装。具有广泛应用潜力。
摘 要
Hi-C 技术可以获得基因组三维构象信息,其被广泛应用于基因组组装。本文发展了GutHi-C技术。如下图摘要所示,该方法高效、快速,可广泛应用于人类、家畜和家禽肠道微生物。开拓了微生物宏基因组Hi-C重要技术手段(名词解释:Hi-C,高通量染色质构象捕获;LB,Luria-Bertani培养液;DPBS,杜氏磷酸盐缓冲液;NGS,下一代测序技术;PCR,聚合酶链反应;QC,质控)。
视频解读
Bilibili:https://www.bilibili.com/video/BV1XusLeJE57/
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
微生物在生态系统中起着关键作用,了解其宏基因组的组织模式对于理解微生物的功能和微生物群体内相互关系具有重要意义。在微生物宏基因组研究中广泛使用的Shotgun技术会产生大量冗余序列,无法在种和菌株水平上进行进一步分类。Hi-C技术在三维(3D)组织模式分析和基因组辅助组装方面具有明显优势,由于技术局限性,在微生物中很少使用,这一优势没能充分利用起来。因此,亟需一种更高效、更易于操作的Hi-C技术,能广泛应用于人类、畜禽或家禽的肠道微生物。本文介绍了一种适合于微生物种群的宏基因组GutHi-C技术(图1A)。本方法进一步优化了实验条件,显著减少了文库浪费和损失,节约了实验试剂(图 S1)。我们还制作了技术操作视频,以促进该技术的学术交流(https://youtu.be/aYEhhRO3eBk)。结果表明,GutHi-C方法的质控参数(如唯一比对率、去重后有效数据比例、有效互作对的比例和顺式相互作用比例)优于以往方法产生的数据。GutHi-C技术还具有良好的可重复性(图 S2)。基于大数据量测序结果显示,本方法表现出明显的Hi-C信号强度,并表现出明显的染色质相互作用结构域,如染色体相互作用域(CIDs)和环结构域。除此之外,在组装方面应用也具有较大应用潜力,GutHi-C数据对经过三代HiFi宏基因组组装数据进行辅助挂载,可以继续提高组装完整的微生物高质量基因组个数(增长比例38.6%)。因此,基于对GutHi-C技术的评估分析,GutHi-C将在动物、人类的肠道微生物和广泛的微生物群落(包括土壤或环境微生物)中得到广泛的应用。
GutHi-C技术的数据评估
在GutHi-C中获得的文库中进行测序(约2千兆碱基的原始数据),并对经过HiC-Pro处理后数据进行了评估。将评估结果(图1B)与Bickhart等人先前发表的Hi-C数据进行对比。GutHi-C表现出明显优势。其数据比对率(唯一比对率)、有效互作对的比例以及去重后有效数据比例与先前文章(均未公开技术细节)中发表的数据具有可比性,这表明本研究所采用的GutHi-C文库构建方法将会具有较多的优势且能获得较好的数据质量。
为了进行更全面的比较,我们选取了兼顾基因组组装质量和Hi-C比对率均较高的前五种微生物基因组,并为它们构建了Hi-C热图(图1C、D)。可以看到,微生物基因组的Hi-C互作信号主要集中在基因组内部,而在基因组外部(与其他微生物基因组互作)几乎检测不到Hi-C信号。我们利用互作频率热图对数据集进行了比较分析。本研究的测试数据包括GUT1-KM1、GUT1-KM2和GUT1-KM3。为了在数据量匹配的情况下进行比较分析,一方面,与本研究的GUT1-KM1进行比较(图1C),我们将对照组数据抽样到约为800万对(图1D)。另一方面,与本研究的GUT1-KM1、GUT1-KM2和GUT1-KM3的组合数据进行比较(图1E),我们将对照组的数据抽样到约为2500万对(图1F)。对比分析显示,在相同数据量条件下,GUT1-KM1(图1C)数据质量显著优于对照组(图1D)。此外,GUT1-KM1、GUT1-KM2和GUT1-KM3组合数据(图1E)的互作频率也超越了对照组(图1F)。即便参考基因组的初始组装质量不佳,GutHi-C技术仍能提供更多的顺式互作(微生物内部或完整Contigs内部)比例。因此,GutHi-C技术在未来的完整宏基因组辅助组装中将展现出巨大的应用潜力。
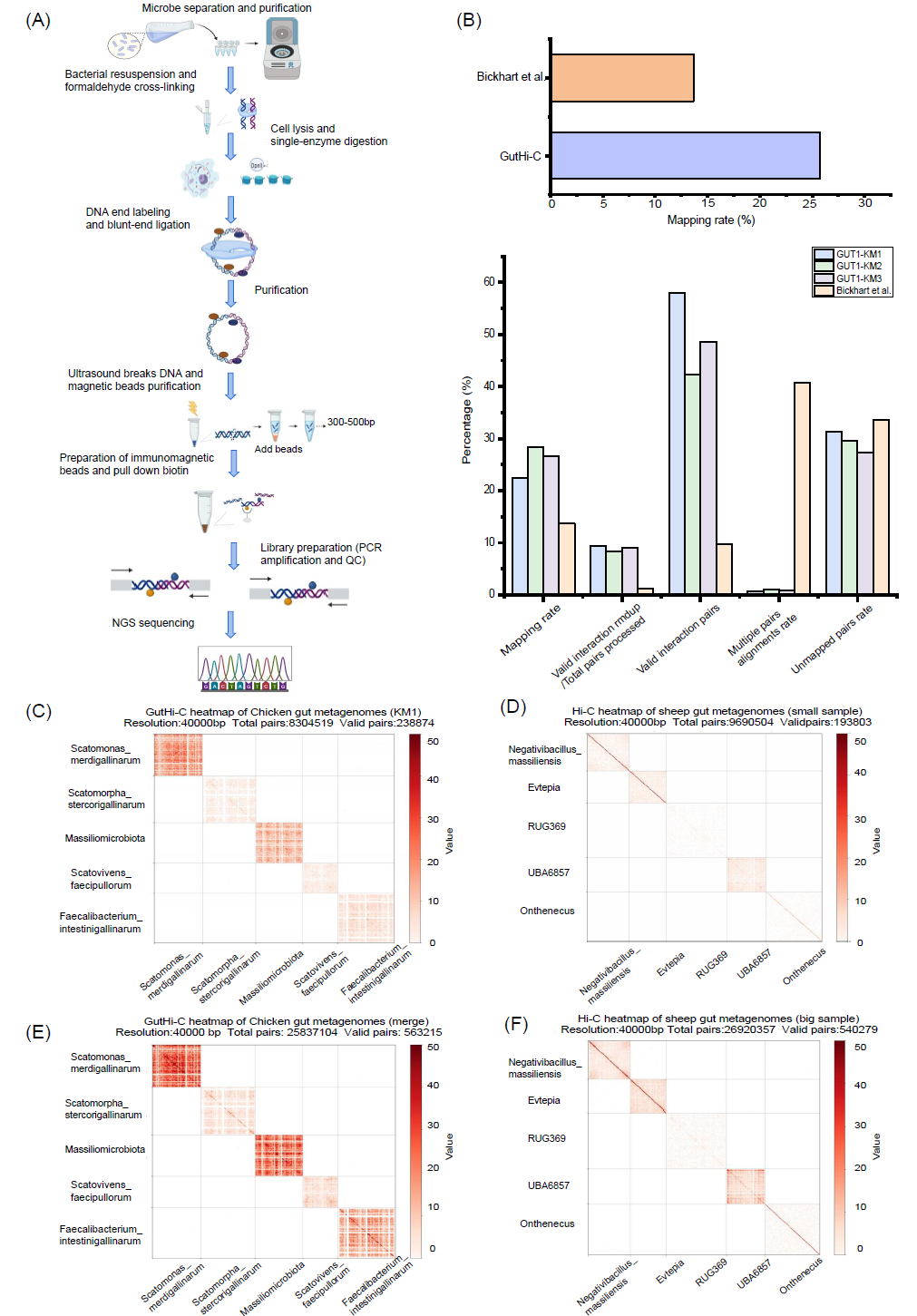
图1. GutHi-C文库的数据评估与结果分析
(A)GutHi-C技术路线图;(B)使用HiC-Pro对GutHi-C和ProxiMeta Hi-C两组数据进行比较评估的结果;(C)鸡肠道微生物宏基因组的Hi-C热图(GUT1-KM1,下载数据约800万对);(D)羊肠道微生物宏基因组的Hi-C热图(下载数据约800万对);(E)鸡肠道宏基因组的Hi-C热图(合并数据约2500万对);(F)羊肠道宏基因组的Hi-C热图(下载数据约2500万对)。
应用GutHi-C揭示微生物宏基因组的三维构象
我们重新采集了实验鸡(岭南黄鸡)的肠道微生物样本,重建了GutHi-C文库,并将这些文库进行大规模测序(获得约100~150千兆碱基的原始数据)。我们分别选择了来自GutHi-C和对照下载数据的前十种单菌进行热图比较。比较结果显示,GutHi-C显示出更明显的信号强度,如图S3A和3B所示。随后,我们聚焦于信号最强的细菌,展示了高分辨率互作频率热图。从20 kb和40 kb分辨率下均可看出,GutHi-C显示出更显著的信号强度(以及局部互作,也称为环)(图2A、B)。此外,单个细菌菌株内存在特定局部区域的互作,如图2C所示,图中黑色实线三角标出的区域代表一个类似拓扑相关结构域(TAD)的强互作频率区域,被称为染色体相互作用结构域(CIDs)。因此,我们的研究结果表明,在GutHi-C方法下的单个细菌中存在代表强相互作用模式的区域。而在类似的大数据量条件下,先前发表的对照组下载数据的Hi-C热图没有显示出有明显的相互作用区存在。
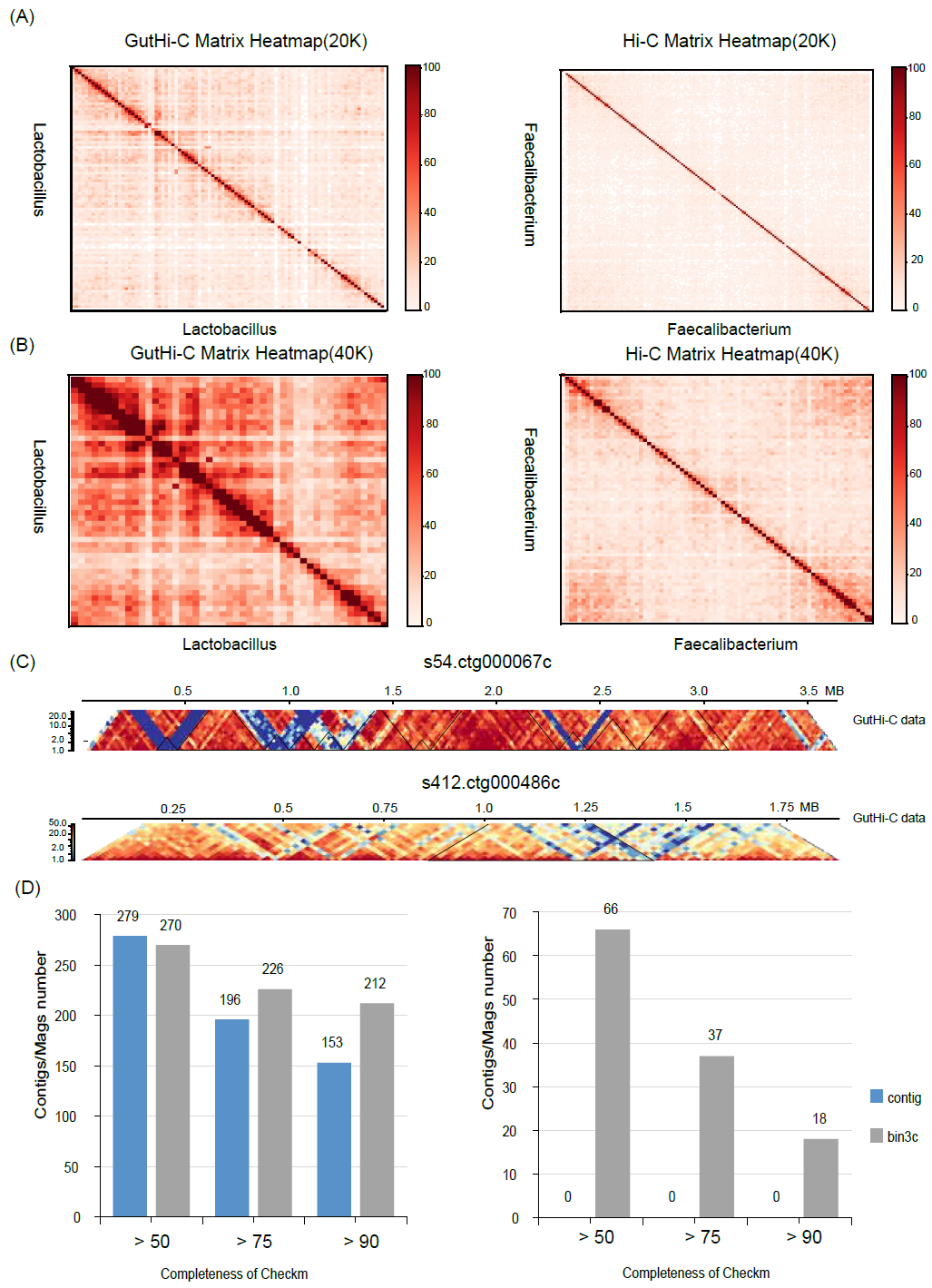
图2. 对微生物宏基因组三维构象的评估分析及初步应用以辅助宏基因组组装
(A)使用GutHi-C技术在20 kb分辨率下的肠道微生物群Hi-C矩阵热图(左),使用ProxiMeta Hi-C技术在20 kb分辨率下的肠道微生物群Hi-C矩阵热图(右);(B)使用GutHi-C技术在40 kb分辨率下的肠道微生物群Hi-C矩阵热图(左),使用ProxiMeta Hi-C技术在40 kb分辨率下的肠道微生物群Hi-C矩阵热图(右);(C)鸡肠道中某些细菌GutHi-C图谱(20 kb分辨率)的染色体相互作用域(CID)及其局部相互作用。(D)基于hifiasm-meta,使用GutHi-C辅助Zhang等人鸡肠道数据集的高保真(HiFi)组装评估(左)。基于spades,使用肠道Hi-C辅助illumina数据集的组装评估。蓝条和灰条分别代表contigs的集合,以及使用bin3c的GutHi-C辅助的宏基因组组装基因组(MAGs)。CheckM2生成的结果显示所有contigs/MAGs的污染程度均小于10%(右)。
应用GutHi-C辅助组装基于HiFi三代测序的完整宏基因组
肠道微生物群的Hi-C技术尚未被广泛应用于辅助微生物完整(成环)宏基因组组装。大多数应用仅限于辅助真核生物的基因组组装或三维构象分析。我们利用GutHi-C文库构建方法对鸡肠道微生物组盲肠样本进行高通量测序分析。我们这里选择了具有较高比对率的样本(样本Gut1)来辅助组装鸡的高保真完整宏基因组,并与Zhang et al.先前发表的鸡肠道宏基因组组装结果进行比较(Zhang et al.之前利用第三代测序HiFi组装获得了可作为参考宏基因组的鸡肠道宏基因组)。在本研究中利用GutHi-C数据辅助HiFi三代测序数据组装鸡的肠道微生物时候,使用了bin3C软件进行分类。同时,我们也对基于Illumina二代测序数据组装的同源样本微生物宏基因组也进行了bin3C分类。如图2D所示,由于GutHi-C数据的质量优良,与高精度的HiFi数据强强结合,我们获得了212个高质量的完整宏基因组(MAGs)(完整性>90且污染参数<10)和226个中到高质量的完整MAGs(完整性>75且污染参数<10)。
与之前的Contig级别组装相比,这标志着高质量基因组的数量增加了38.6%。然而,中等质量的MAGs(完整性>50且污染参数<10)数量有所减少。这表明利用GutHi-C数据可以对低质量或中等质量的Contigs进行分类,从而提升HiFi得到的高质量宏基因组的组装质量。图2D也展示了,GutHi-C在提升二代shotgun测序(NGS)组装得到的宏基因组的组装质量方面的显著效果。在使用illumina TruSeq Shotgun测序进行contigs的组装时,没有能达到中等质量的MAGs(0个MAGs),并且中等到高质量MAGs是0个,高质量的MAGs也是0个。然而,借助相应的GutHi-C数据集进行辅助组装,我们获得了66个中等质量的MAGs(完整性>50且污染参数<10),37个中等到高质量的MAGs(完整性>75且污染参数<10)以及18个高质量的MAGs(完整性>75且污染参数<10)。
讨 论
GutHi-C的技术优势本文按技术步骤的顺序进行讨论。GutHi-C使用液氮研磨和溶菌酶处理实现微生物裂解。这样,微生物细胞壁得以最大程度地渗透,使得试剂可以充分接触DNA。其有利于后续的限制性内切酶对微生物基因组的彻底切割(片段化),并且使连接酶可以在核区有效反应并产生高质量的“邻近连接”,从而确保后续文库构建步骤的高效率。这种方法可以得到更高的有效互作对比例和更高的去重后有效数据比例。此外,仅使用溶菌酶处理进行微生物裂解这一方式,能够减少文库损失并显著提高后续步骤的DNA浓度,使其在微生物群体中微生物数量较少时产生优势。因为液氮研磨可能会损失少量的微生物样品。因此,当微生物数量极小时,可以单独进行溶菌酶裂解以获得所需的较高DNA浓度样本。在GutHi-C的文库构建过程中,引入了“原位Hi-C框架”,这能够保持核区域的原始微环境,使邻近连接在核区域内最大程度地进行,提高了连接效率,并且相比于传统Hi-C降低了背景噪音。此外,本研究中使用的邻近连接反应溶液包含易于获取的重组白蛋白,它替代了现有技术中的BSA并发挥了相同的作用。在本研究中,生物素作为平末端标记,但其用量仅为常规原位Hi-C系统所需的一半,仍然保持了良好的效果。换句话说,这不仅能维持或增强了原有效果,还可以通过将技术步骤中最昂贵的生物素标记试剂的使用量减半,降低实验成本。GutHi-C采用T1免疫磁珠进行文库捕获,T1磁珠的用量已经从150 μL 减少到50 μL,但仍保持了文库构建效率(图S1)。这有助于实验成本的持续降低。此外,该方法将嵌合互作DNA富集步骤置于NGS建库加A步骤和加接头步骤之前,从而减少了试剂消耗。最后,在DNA正式扩增前进行聚合酶链反应质量控制(PCR-QC)测试,可以获得最佳的扩增条件,提高GutHi-C文库的制备比例,避免试剂浪费。更重要的是,它能显著减少文库损失。大量的条件、步骤调整测试获得的以上这些优势是GutHi-C结果优于现有技术的原因。另外,通过设置不同的实验变量进行比较,结果表明GutHi-C具有良好的重复性(图S2)
鉴于当前关于鸡肠道微生物宏基因组Hi-C数据的文献报道甚少。一方面,我们无法下载到对应的鸡的对照组数据;另一方面,可用的、方法成熟的微生物宏基因组Hi-C或试剂盒寥寥无几。例如,国际上现有的一款微生物Hi-C方法细节及其试剂盒在国内难以获取(作者在新闻稿注:基于现有文献数据,例如Joshua等人,G3 (Bethesda),2014;Ivan等人,bioRxiv,2017;Stewart等人,Nat Commun, 2018;Stalder等人,ISME J, 2019;Bickhart等人,Nat Biotechnol, 2022;Gounot等人,Nat Commun, Genome Biology, 2022;发表的微生物Hi-C上述文献均来自ProxiMeta Hi-C方法)。因此,我们下载了发表的ProxiMeta Hi-C数据作为对照组,例如由ProxiMeta Hi-C技术准备的绵羊和牛的肠道微生物宏基因组Hi-C数据(作者在新闻稿注:这里不排除如果能将该ProxiMeta Hi-C试剂盒应用在鸡肠道微生物组也能得到与GutHi-C相当的结果,作者也期待未来发表的文章中会出现该类研究,本研究仅为证明GutHi-C的可行性和高效性,我们发展了一款优秀的国产替代技术)。基于比较这些数据,GutHi-C相较于现有Hi-C技术展现出更强的Hi-C信号强度,并呈现了较强的染色质互作域(图2A-C,图S3)。同时,在补充材料中,我们补充并全面评估了GutHi-C技术的实验结果及其优越性。为了更好的拓展国内领域的学术交流,我们也与从Phase Genomics官网下载的ProxiMeta试剂盒手册中的实验程序进行了比较(表S1)(作者在新闻稿注:我们了解到ProxiMeta试剂盒成分保密,步骤不透明,方法无法标准化,仅对比了模块化的步骤差异,仅供学术交流)。
Gut Hi-C的一个重要应用方向是完整宏基因组辅助组装。ProxiMeta Hi-C在组装牛瘤胃微生物的宏基因组时,减少了所需的测序量。在这两项研究中,他们分别采用了Hi-C与二代Illumina TruSeq Shotgun测序以及Hi-C与三代PacBio RS SMRT测序相结合的组装策略。众所周知,Illumina Shotgun的读长通常只有150~250 bp,而PacBio SMRT的错误率比较高(约15%)。因此,这可能导致成倍的重复序列的出现和因其复杂性而丢失无法完全组装的区域。本研究具有另一重要意义,就是我们展示了GutHi-C技术在完整宏基因组组装中的两个良好的应用实例。一方面,证实了Hi-C技术与二代illumina测序结合可以显著增加了环形微生物基因组的数量。另一方面,我们验证了利用Hi-C辅助HiFi读长的高质量组装,以获得更完整、高保真的单菌分辨率水平微生物组环状完整宏基因组的可行性。
因此,我们预测GutHi-C技术可能具有与当前其他技术相媲美的影响力和应用范围。
另外,GutHi-C技术成果转化或横向项目合作,请联系kongsiyuan@caas.cn,或中国农业科学院深圳农业基因组研究所产业发展处(电话:0755-28398803)。

引文格式:
Lu, Y.-X., Yang, J.-B., Li, C.-Y., Tian, Y.-H., Chang, R.-R., Kong, D.-S., Yang, S.-L., Wang, Y.-F., Zhang, Y.-B., Zhu, X.-S., Pan, W.-H. and Kong, S.-Y. (2024). "Efficient and easy-to-use capturing three-dimensional metagenome interactions with GutHi-C." iMeta e227. https://doi.org/10.1002/imt2.227
作者简介

卢宇曦(第一作者)
● 河南大学与中国农业科学院深圳农业基因组研究所联合培养生物学硕士。
● 主要研究领域为动物三维基因组学,研究方向为动物基因组学技术;主要成果发表在iMeta、Journal of Integrative Agriculture、中国畜禽种业期刊。

李晨莹(第一作者)
● 青岛农业大学动科院畜牧专业,与中国农业科学院深圳农业基因组研究所联合培养在读硕士。
● 研究方向:动物繁殖育种;要成果发表在iMeta、Journal of Integrative Agriculture、中国畜禽种业期刊。

杨金宝(第一作者)
● 中国农业科学院深圳农业基因组研究所与华中农业大学联合培养博士生。
● 主要研究领域为基因组学与生物信息学算法,研究方向为基因组及宏基因组组装算法的开发、三维基因组分析、T2T基因组组装等;主要成果发表在Genome Research、iMeta、Horticulture Research等期刊。

孔思远(通讯作者)
● 中国农业科学院(深圳)农业基因组所Pre-PI课题组长,特聘副研究员,中国农业科学院深圳研究生院教师,华中农业大学、广西大学、青岛农大等联培高校硕士生导师,农业农村部畜禽生物组学重点实验室成员。国家“博士后创新人才支持计划”、深圳市优秀科技创新人才培养计划(博启/青年)入选者,中国农业科学院“优秀博士后”、深圳市“优秀博士后”获得者。
● 围绕畜禽基因组前沿育种理念,从事高精度多组学技术研发及应用,为动物分子遗传育种等畜牧学基础领域解析提供新的组学技术;结合基因和细胞工程挖掘育种用优异基因和调控元件,精准解析畜禽和模式动物的转录调控、营养调控分子机制。相关研究以第一或通讯作者在Nucleic Acids Research(2篇,IF=16.6)和 iMeta(IF=23.4)等期刊发表文章 14 篇(累积IF 87.29)。授权国家发明专利7项(前两位)。主持国家自然科学基金青年项目、中国博士后基金面上项目和广东省自然科学基金面上项目等 8 项;担任中国畜牧兽医学会高级会员,中国细胞生物学学会会员,广东省基础与应用基础研究基金项目评审专家,广东高校科技成果转化专家库入库专家,深圳市科技创新局在库专家;iMetaOmics期刊执行副主编,JoVE(Journal of Visualized Experiments)客座主编;担任Animal Research and One Health,Agriculture Communications,Animals and Zoonose,Advanced Biotechnology,中国畜禽种业等优秀期刊青年编委;担任生物技术通报,农业生物技术学报,Journal of Integrative Agriculture审稿专家。曾获得吴常信动物遗传育种优秀论文奖、全国动物遗传育种学术讨论会优秀论文特等奖、中国农业科学院“优秀教师团队”教师教学奖等奖励。

潘玮华(通讯作者)
● 研究员,博士生导师,深圳市海外高层次人才。
● 2019年获美国加州大学河滨分校计算机科学博士学位和统计学硕士学位。2019-2020年在美国卡内基梅隆大学计算机学院计算生物学系担任Lane Fellow(博士后)。长期从事基因组学,尤其是基因组序列分析相关的生物信息算法研究。主要成果发表在顶级会议RECOMB、ISMB和专业期刊National Science Review、Genome Research、Molecular Plant、Bioinformatics、Genomics Proteomics & Bioinformatics、Plant Physiology等。主持国家自然科学基金、深圳市优秀科技创新人才培养项目等科研项目。担任中国生物信息学学会(筹)生物信息算法专委会委员、中国生物工程学会计算生物学与生物信息学专委会委员等。担任Frontiers in Plant Science和Genes等期刊客座编辑。

朱秀生(通讯作者)
● 博士后,研究领域为功能基因组学。
● 长期专注于非编码区功能调控元件高通量鉴定技术的开发及其机制研究,2023年10月入站以来,获得深圳市大鹏新区“鹏程计划”科技人才项目和中国农业科学院“优农计划”项目重点资助,以第一作者或通讯作者(含共同)在Nucleic Acids Research,iMeta ,Computational and Structural Biotechnology Journal,Genes & Diseases等期刊发表SCI论文6篇,总影响因子约60分,授权国家发明专利4项。
更多推荐
(▼ 点击跳转)
iMeta | 引用13000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期

3卷1期

2卷2期封底

2卷4期封底

3卷2期

3卷3期

3卷3期封底
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百千华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 20)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!发行后相继被Google Scholar、ESCI、PubMed、DOAJ、Scopus等数据库收录!2024年6月获得首个影响因子23.7,位列全球SCI期刊前千分之五(107/21848),微生物学科2/161,仅低于Nature Reviews,同学科研究类期刊全球第一,中国大陆11/514!
“iMetaOmics” 是“iMeta” 子刊,主编由中国科学院北京生命科学研究院赵方庆研究员和香港中文大学于君教授担任,定位IF>10的高水平综合期刊,欢迎投稿!
联系我们
iMeta主页:
http://www.imeta.science
姊妹刊iMetaOmics主页:
http://www.imeta.science/imetaomics/
出版社iMeta主页:
https://onlinelibrary.wiley.com/journal/2770596x
出版社iMetaOmics主页:
https://onlinelibrary.wiley.com/journal/29969514
iMeta投稿:
https://wiley.atyponrex.com/journal/IMT2
iMetaOmics投稿:
https://wiley.atyponrex.com/journal/IMO2
邮箱:
office@imeta.science


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









