






点击蓝字 关注我们
整合全基因组和全转录组关联分析揭示牛瘤胃甲烷生成中的宿主-微生物相互作用

研究论文
● 期刊:iMeta(IF 23.7)
● 原文链接DOI: https://doi.org/10.1002/imt2.234
● 2024年9月3日,西北农林科技大学王禹、武圣儒和浙江大学李福勇等在iMeta在线发表了题为“Integrating genome- and transcriptome-wide association studies to uncover the host-microbiome interactions in bovine rumen methanogenesis”的文章。
● 本研究使用大规模配对样本的基因组、转录组和微生物组数据,系统评估了牛瘤胃微生物组成的宿主遗传基础,在基因表达水平上建立起宿主与微生物之间的互作关系,为从遗传调控和微生物干预策略减缓反刍动物甲烷排放提供了重要参考。
● 第一作者:王伟、魏振宇、李卓辉
● 通讯作者:王禹(wang_yu@nwafu.edu.cn)、武圣儒(wushengru2013@nwafu.edu.cn)、李福勇(fuyong@zju.edu.cn)
● 合作作者:任建荣、宋颜亮、许婧怡、刘安国、李鑫妹、李曼曼、樊慧梅、金良梁、Zhannur Niyazbekova(张诺)、王文、高元鹏、姜雨、姚军虎
● 主要单位:西北农林科技大学动物科技学院、浙江大学动物科学学院、西北工业大学生态环境学院、西北农林科技大学家畜生物学重点实验室
亮 点
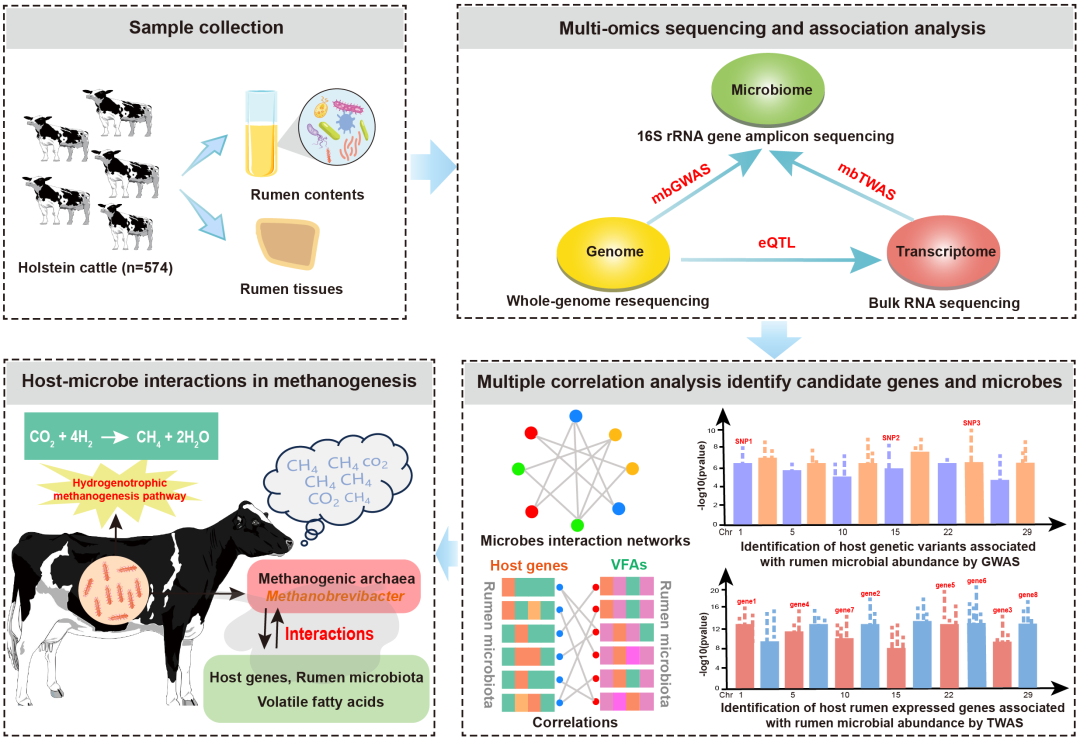
● 利用大规模配对样本,揭示了宿主遗传变异和瘤胃基因表达对荷斯坦牛瘤胃微生物菌群的影响;
● 宿主遗传因素平均解释了大约28%的微生物丰度差异,而瘤胃基因表达能够解释约43%;
● TWAS在检测直接效应器官基因表达与复杂性状表型关联方面具有突出优势;
● 牛瘤胃甲烷生成过程中的宿主-微生物相互作用可能与底物氢的代谢和转运有关。
摘 要
反刍动物的瘤胃微生物能够产生生物甲烷。然而,宿主遗传在改变瘤胃微生物菌群介导的甲烷排放中的作用仍然是一个谜,这严重阻碍了这种臭名昭著的温室气体的排放控制。在本研究中,我们通过对574头中国荷斯坦牛的基因组、瘤胃转录组和微生物组数据进行全基因组和全转录组关联研究,系统揭示了瘤胃微生物组成的宿主遗传基础。遗传力评估发现,约70%的微生物类群具有显著遗传力,但通过全基因组关联研究(GWAS)方法仅鉴定出与22个微生物类群显著关联的43个遗传变异。相比之下,全转录组关联研究(TWAS)方法检测到28,260个显著的基因-菌群关联对,涵盖了210个分类群和4652个非冗余基因。宿主遗传因素平均解释了大约28%的微生物丰度差异,而瘤胃基因表达能够解释约43%。此外,我们强调TWAS在检测直接效应器官的基因表达和表型性状关联方面具有很强的优势。对于产甲烷古菌,GWAS只检测到1个显著关联位点,而TWAS获得了1703个显著关联的宿主基因。通过结合这些宿主TWAS基因、瘤胃菌群和挥发性脂肪酸之间的多重相关性分析,我们发现底物氢代谢是联系牛瘤胃甲烷生成中宿主-微生物相互作用的重要途径。总之,这些发现为通过遗传调控和微生物干预策略减缓反刍动物甲烷排放提供了有价值的指导。
视频解读
Bilibili:https://www.bilibili.com/video/BV19iHoecEFd/
Youtube:https://youtu.be/zh2aV3kTAdQ
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
甲烷作为六大温室气体之一,其对全球变暖的影响仅次于二氧化碳。减少畜牧业生产中的甲烷排放对于中国实现碳中和目标至关重要。反刍动物的瘤胃微生物菌群能够产生甲烷,其贡献了人为甲烷排放总量的约18%。牛作为主要的家养反刍动物,贡献了大部分畜牧生产甲烷排放,这归因于其强大的瘤胃微生物发酵功能。甲烷的产生与瘤胃中产甲烷古菌的丰度密切相关,其中大部分产甲烷古菌主要来自甲烷短杆菌属(Methanobrevibacter)。例如Methanobrevibacter gottschalkii、Methanobrevibacter smithii、Methanobrevibacter boviskoreani、Methanobrevibacter millerae和Methanobrevibacter thaurei等。这些都是常见的氢营养型产甲烷古菌,通常利用微生物发酵产生的氢气和二氧化碳作为底物产生甲烷。动物肠道甲烷排放除了对环境产生负面影响外,还会导致2~12%的宿主能量摄入损失。因此,寻找调控瘤胃微生物的方法来减缓反刍动物的甲烷排放一直是迫切需要的。
肠道菌群受饮食、宿主、环境等多种因素的影响。宿主衍生分子对肠道菌群的调控为理解宿主-微生物相互作用提供了新的视角。越来越多的研究强调了宿主遗传对人类和动物肠道微生物群的重要作用。肠道菌群的遗传力估计有助于了解宿主遗传因素解释微生物丰度变化的比例。在人类TwinsUK群体研究中,发现粪便样本中5.3%~8.8%的细菌分类群的遗传力大于0.2。反刍动物的瘤胃微生物菌群似乎受宿主遗传因素的影响更大,大约34%的微生物类群和64%的核心属分别在709头肉牛和1150只绵羊的大队列研究中发现具有显著的遗传力。近年来,多项研究利用微生物组作为复杂表型,通过全基因组关联研究(GWAS)鉴定宿主遗传变异与微生物丰度变化之间的关系。然而,尽管有报道称甲烷排放量和产甲烷菌丰度也具有中等遗传力,但通过GWAS方法进行的多项研究中并未发现瘤胃产甲烷古菌相关的主效信号位点。与GWAS相比,全转录组关联研究(TWAS)被用于鉴定基因表达与表型之间的关联关系,这对于解释复杂表型的遗传机制以及提供基因水平的关联具有重要价值。因此,通过TWAS方法建立宿主瘤胃表达基因与瘤胃产甲烷菌之间的相关性,将成为揭示宿主调控甲烷排放的一种全新策略。然而,据我们所知,整合大规模配对样本的GWAS和TWAS分析尚未用于动物肠道微生物研究,特别是反刍动物瘤胃微生物产甲烷领域。
在本研究中,我们旨在建立瘤胃甲烷生成中宿主与微生物之间的潜在关系。我们猜测,与宿主遗传因素相比,瘤胃基因与瘤胃微生物之间的关系更为直接,宿主通过驱动瘤胃基因表达来影响微生物变化,从而参与甲烷排放调控。为了验证这些假设,我们对574头荷斯坦牛配对的基因组、转录组和微生物组数据进行了微生物全基因组关联研究(mbGWAS)和全转录组关联研究(mbTWAS),以确定影响瘤胃微生物群的宿主遗传变异和瘤胃表达基因(图1A)。这些工作将为通过遗传调控瘤胃菌群来减缓反刍动物甲烷排放的可能性做出重要探索,并拓宽了我们对瘤胃甲烷生成中宿主-微生物相互作用潜在机制的认识。
结 果
大队列荷斯坦牛瘤胃微生物菌群特征
本研究对574头成年中国荷斯坦牛的瘤胃内容物样本进行了16S rRNA基因扩增子测序。经过严格质量过滤,共获得了32,622,851条高质量的细菌和古菌序列。平均每个样本分别获得35,336条和22,234条细菌和古菌有效序列(图S1A和S1D)。将序列以99%相似性聚类为扩增序列变体(ASV)。我们分别获得了5974和3588个细菌和古菌的ASVs(表S1和S2),平均每个样本中ASVs分别为1175和65个(图S1B和S1E)。经分类学注释,保留至少在20%样本中检出的分类群,细菌在五级分类水平上共检测到269个分类群(包括13个门、19个纲、33个目、60个科和144个属),古菌在六级分类水平上共检测到48个分类群(包括3个门、4个纲、4个目、4个科、8个属和25个种)(图S1C、S1F和表S3)。细菌优势菌门为拟杆菌门(56.4%)、厚壁菌门(34.5%)和变形菌门(3.3%)(图1B);优势菌属为Prevotella(17.7%)、Rikenellaceae_RC9_gut_group(16.9%)和Bacteroidales_F082(7.6%)(图1C)。古菌优势属门为广古菌门(64.4%)和Thermoplasmatota(34.7%)(图1D);优势菌种为Methanobrevibacter gottschalkii(26.8%)、Methanobrevibacter ruminantium(24.2%)和Methanomassiliicoccus luminyensis(15.3%)(图1E)。采用α和β多样性对微生物群落组成结构和多样性进行分析,结果显示细菌的微生物丰富度高于古菌(图1F和1G),这些研究个体具有相似的微生物组成和群落结构(图1H和1I)。此外,为了进一步探究瘤胃微生物群落之间的互作网络,我们使用Spearman相关性分析构建了属水平上的微生物相关网络,鉴定出58个强相关类群(|r| > 0.5,矫正p < 0.05)(图1J)。其中,Oscillospiraceae_NK4A214_group、Christensenellaceae_R-7_group、Lachnospiraceae_NK3A20_group、Ruminococcus、Prevotellaceae_UCG-003等菌属在互作网络中具有较高的连接性且存在于几乎所有个体中,这些分类群可能代表了荷斯坦牛的瘤胃核心菌群。我们还使用PICRUSt2软件预测了所有ASV序列的16S rRNA基因的功能通路。结果显示,普雷沃氏菌属与L-鼠李糖降解有关;Christensenellaceae_R-7_group与(R, R)-丁二醇生物合成和肌醇降解的超途径有关。甲烷短杆菌属与多个甲烷生成相关的功能途径呈正相关,如辅酶M生物合成、F420生物合成以及从氢气(H2)和二氧化碳(CO2)生成甲烷(CH4)等(图S2)。
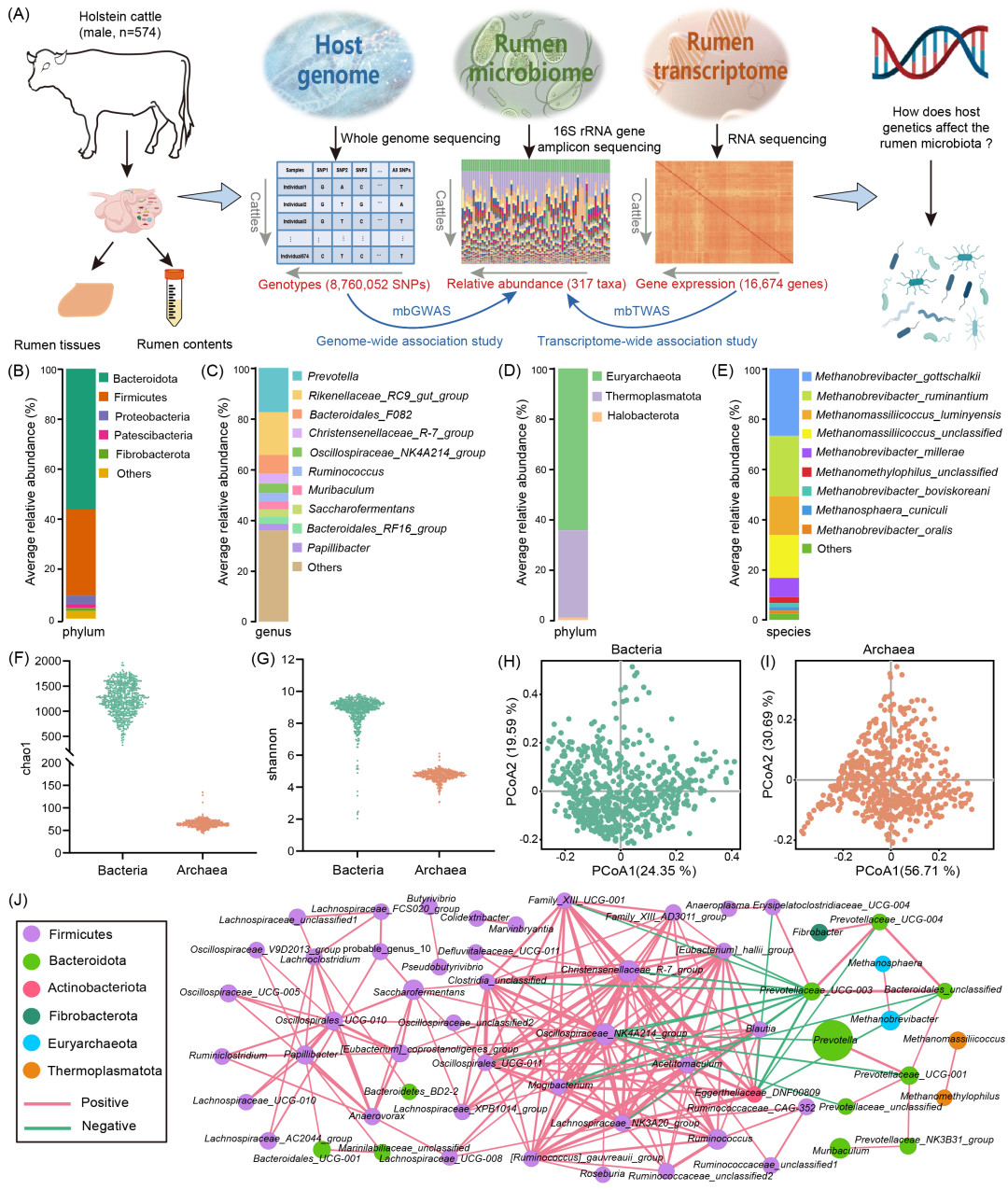
图1. 研究设计及瘤胃微生物组成和群落结构
(A)工作流程和技术路线图;(B)和(C)细菌在门和属水平上分类群的组成和丰度;(D)和(E)古菌在门和种水平上分类群的组成和丰度;(F)和(G)利用chao1和shannon指数比较细菌和古菌的α多样性差异;(H)和(I)细菌和古菌在属水平上基于Bray-Curtis距离的β多样性PCoA图。(J)属水平分类群的互作网络;仅展示相关系数大于0.5或小于-0.5且矫正p值小于0.05的类群,节点大小代表平均相对丰度。
宿主遗传因素影响瘤胃微生物
对574头荷斯坦牛个体进行全基因组测序,每个个体的平均测序深度约为7×,共产生10.5 Tb的原始数据。经过严格的质量控制和变异过滤,最终获得了8,760,052个全基因组遗传变异(SNPs)。使用基于这些SNPs的主成分分析(PCA)来评估群体遗传结构(图S3A)。通过遗传变异在29条常染色体上的分布(图S3B)和基因注释显示,大多数SNPs存在于基因间区(56.3%)或内含子区(35.9%)(图S3C和表S4)。为了研究宿主遗传是否影响瘤胃微生物群,使用Mantel检验对宿主遗传亲缘关系矩阵和菌群Bray-Curtis相似性矩阵进行了相关性分析。鉴于大多数个体对表现出较低程度的遗传亲缘关系(图2A),我们随机选取了500对遗传距离较远和较近的个体对分别进行相关性计算,其相关系数分别为0.06和0.11(图2B和2C)。这些结果表明,宿主遗传亲缘关系与微生物菌群相似性之间存在弱正相关性。随后,我们评估了317个微生物特征的遗传力(h2),包括171个丰度连续性状和146个存在/缺失二元性状分类群(图2D和表S5)。我们发现,宿主遗传因素平均解释了约28%的微生物丰度差异,约70%(223/317)的分类群具有显著的遗传力(似然比检验,p < 0.05)。古菌的平均遗传力(0.41±0.13)高于细菌(0.36±0.14)(图2E)。在这些可遗传的细菌分类群中,50%以上属于厚壁菌门(图2F),可遗传的古菌主要分布在Thermoplasmatota和Euryarchaeota门(图2G),例如Oscillospiraceae_UCG-005(h2 = 0.77)、Christensenellaceae_R-7_group(h2 = 0.46)和Methanobrevibacter_gottschalkii(h2 = 0.47)(图2H)。从以上结果可以看出,宿主遗传在一定程度上影响着瘤胃微生物群,瘤胃核心菌群的遗传力可能几乎是普遍存在的。
为了深入了解宿主加性遗传对瘤胃微生物丰度表型的影响,我们对574头牛的8,760,052个常染色体遗传变异与223个可遗传分类群表型进行了全基因组关联研究(GWAS)。在GWAS分析中,没有发现检验统计量膨胀,(基因组膨胀系数(λGC)中位数为1.009),说明模型拟合良好(图S4)。我们在全基因组显著阈值5.71×10-9下,鉴定到43个SNPs与22个菌群丰度相关,位于17个独立基因座位(图2I和表S6)。例如,在21号染色体上42.9 Mb区域的显著信号位点与Lachnospiraceae_ND3007_group丰度相关,其中最显著的SNP(21:422903221,p = 1.25×10-9)定位于AKAP6基因的内含子区(图S5A−D)。此外,在我们的研究中,我们同时测定了瘤胃组织的RNA-seq数据,这些大规模配对的基因组和转录组数据为检测瘤胃中的cis-eQTLs提供了前所未有的机会。利用FastQTL软件中的线性回归模型,对454个个体的8,760,052个SNPs和16,674个基因进行了cis-eQTL鉴定。共检测到499,502个显著的变异-基因关系对,包括1619个独立的eGenes和416,109个独立的eSNPs。为了推断宿主遗传变异是否通过影响瘤胃基因表达来影响瘤胃微生物。我们使用coloc软件对cis-eQTLs和GWAS显著位点进行了共定位分析,从770个共享SNPs中发现了86个可能的共定位位点(表S7)。例如,我们发现瘤胃POLM基因的一个cis-eQTL与Lachnospiraceae_FCS020_group丰度性状的mbGWAS位点共定位(图S6A−D)。综上所述,这些结果表明宿主遗传可以影响和调控瘤胃微生物。然而,宿主遗传变异和菌群丰度关联背后的潜在遗传机制以及直接效应器官的影响尚不清楚。因此,建立瘤胃效应器官基因表达与瘤胃微生物群之间的相互作用就显得尤为重要和迫切。
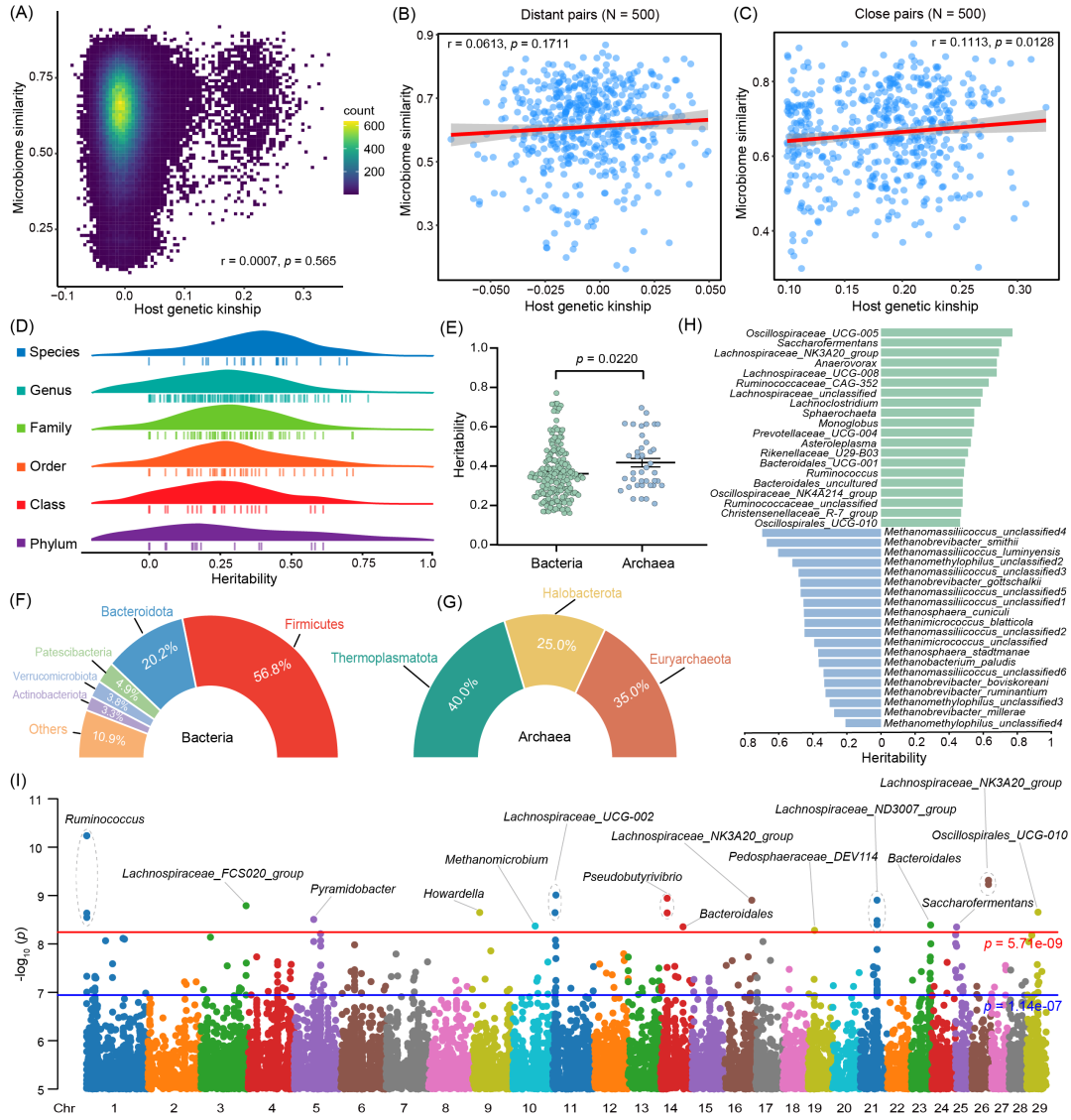
图2. 宿主遗传与瘤胃微生物菌群关系研究
(A)宿主遗传亲缘关系与菌群Bray-Curtis相似性之间的相关关系;随机选择500对遗传上距离较远(B)和较近(C)个体计算相关性;(D)对317个单一分类群性状的遗传力估计;(E)细菌和古菌分类群的遗传力大小比较;(F)和(G)细菌和古菌可遗传类群在不同门水平上的占比分布;(H)细菌和古菌中遗传力排名前20的类群;(I)223个具有显著遗传力瘤胃微生物的全基因组关联分析曼哈顿整合图。曼哈顿图显示了每个SNP的最小结果p值。全基因组显著性阈值(p = 5.71×10-9)用红线表示,建议性显著阈值(p = 1.14×10-7)用蓝线表示。
瘤胃微生物的TWAS图谱
鉴于大多数GWAS显著关联位点通常难以解释,在过去几年中,研究人员投入大量的努力通过使用GTEx汇总数据预测的基因表达,用TWAS方法来识别性状相关的基因。在本研究中,我们收集了具有动态对应的基因表达和微生物丰度数据。因此,我们首次基于16,674个基因与317个分类群的关联构建了瘤胃微生物群的TWAS图谱。我们检测到28,260个显著的基因-菌群关联对(p < 3×10-6),其中包括16,423个正相关和11,837个负相关,涵盖了210个分类群和4652个非冗余基因(图3A和图S7)。我们发现使用TWAS比使用GWAS和cis-eQTL能发现更多关联基因,三种方法仅存在53个共享基因(图S8)。因此,我们推测瘤胃基因表达的动态变化与微生物丰度之间存在更直接的关系。为了进一步证实这一点,我们评估了基因表达关系矩阵与微生物相似性矩阵之间的相关性。结果显示,基因表达与菌群相似性之间的相关性(r = 0.039,p = 0.001)强于宿主遗传亲缘关系与菌群相似性之间的相关性(r = 0.003,p = 0.495),并且这种效应在TWAS基因中更为明显(r = 0.060,p = 0.001)(图3B−D)。随后,我们基于类似于遗传力的方法评估了所有分类群性状的“表达力”,发现瘤胃基因表达平均解释了约43%的微生物组差异(表S8)。遗传力和表达力之间呈显著正相关(r = 0.72,p < 2.2e-16)(图3E);表达力显著高于遗传力(图3F)。此外,TWAS相关特征显示,关联到TWAS基因最多的细菌和古菌分别为[Eubacterium]_hallii_group和Methanomicrobium(图3G)。细菌和古菌类群在TWAS检测基因数量上无显著差异(图3H)。可遗传的瘤胃菌群(h2 > 0.2)具有更多的TWAS基因(图3I)。与瘤胃随机菌群相比,瘤胃核心菌群(检出率> 80%)具有更高的遗传力、表达力和更多的TWAS基因(图3J)。为了阐明瘤胃微生物TWAS关联基因的功能,我们比较了细菌和古菌TWAS基因功能富集的差异。发现细菌和古菌共享1294个TWAS基因(图3K),这些基因显著富集在Rap1信号通路、Calclum信号通路和代谢通路中(图3L)。细菌特异性基因主要在代谢、免疫和病原微生物感染通路中显著富集(图3M)。古菌特异性基因主要富集于长寿调节通路、丙酮酸代谢和糖酵解/糖异生相关的通路(图3N)。此外,鉴于我们是首次对单个组织进行较大样本量的TWAS分析。因此,我们评估了不同样本量(100、200、300、400和全部454)对TWAS分析结果的影响。结果表明,随着样本量的增加,TWAS检测到的基因数量逐渐增加。例如,当使用100个样本时,Christensenellaceae_R-7_group只有2个显著相关基因,而当样本量增加到200个时,则有49个显著相关基因。在样本数大于200后,能捕获到显著TWAS基因的分类群数量逐渐趋于稳定(图S9A−C)。基于此,我们建议在进行TWAS分析时,样本量应至少大于200个,以便获得理想的结果。
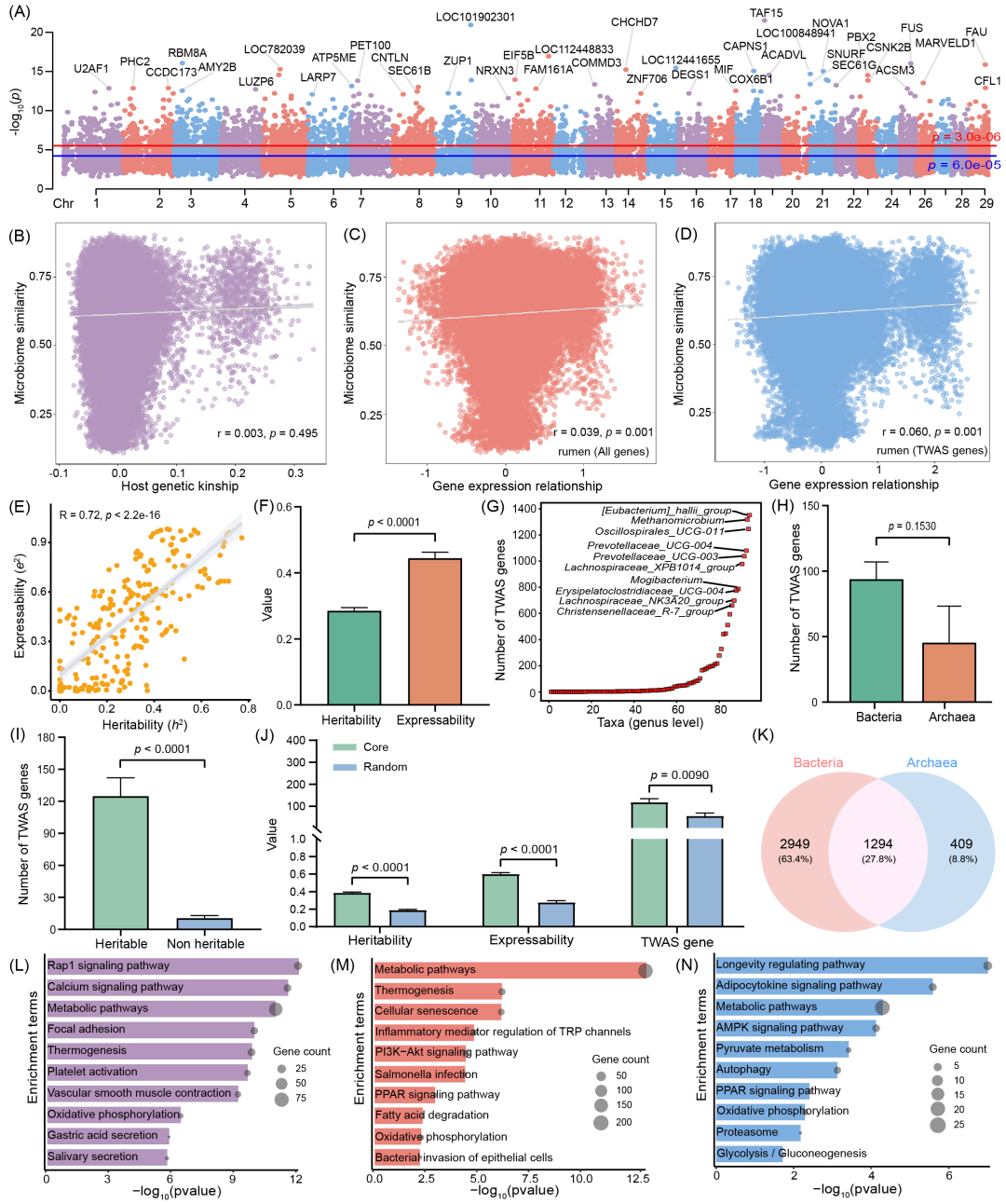
图3. 瘤胃微生物TWAS图谱特征
(A)瘤胃基因表达与瘤胃微生物丰度表型的全转录组关联研究;曼哈顿图显示了每个基因的最小p值结果;(B)454个体中宿主遗传亲缘关系与微生物菌群相似性之间的相关性;分别在所有基因(C)和TWAS显著基因(D)中构建基因表达相关关系矩阵与微生物相似性矩阵之间的相关性;(E)遗传力与表达力之间的Spearman相关性分析;(F)遗传力与表达力差异比较;(G)属水平分类群TWAS基因数量排序图;(H)细菌和古菌中分类群TWAS基因数量的比较;(I)可遗传类群(h2 > 0.2)与不可遗传类群(h2 ≤ 0.2)间TWAS基因数目比较;(J)瘤胃核心菌群(检出率 > 80%)和随机菌群(检出率20~80%)在遗传力、表达性和TWAS基因数量方面的差异比较;(K)瘤胃细菌和古菌之间共享的TWAS基因;(L−N)细菌和古菌共享TWAS基因、细菌特异TWAS基因及古菌特异TWAS基因的功能富集分析。组间比较,结果用平均值±标准误表示,p值小于0.05认为组间差异显著。
整合GWAS和TWAS揭示瘤胃古菌的宿主关联特征
为了进一步研究宿主遗传变异和瘤胃基因表达与瘤胃古菌丰度的关系,我们对48个古菌类群性状进行了GWAS和TWAS分析。根据p < 1×10-5的显著性阈值,使用GWAS方法共鉴定出3049个SNPs,这些SNPs注释到1274个邻近基因(图4A)。于此同时,在相同阈值条件下,利用TWAS方法获得了3201个显著的基因-分类群关联对,涵盖2411个基因和37个菌群(图4B)。通过比较这两个基因集,我们发现GWAS和TWAS方法仅捕获到3.3%的共享基因(图4C)。值得注意的是,在Bonferroni校正阈值下,采用GWAS方法仅存在一个显著的SNP位点(p < 5.71×10-9)与Methanomicrobium丰度相关(图4D)。相比之下,TWAS方法共鉴定出1703个显著关联基因(p < 3×10-6)(图4E)。这些结果提示,对于一些复杂的表型,如微生物组丰度,受宿主微效多基因调控,仅靠GWAS方法可能无法找到有效的遗传标记。与此互补,通过TWAS方法建立基因表达与复杂表型之间的相关性,可以在基因水平上提供更多的分子标记,已成为揭示复杂表型分子机制的重要工具。因此,我们鉴定到的这些瘤胃古菌相关的宿主基因为从宿主-微生物互作角度解决甲烷排放问题提供了全新的机会。
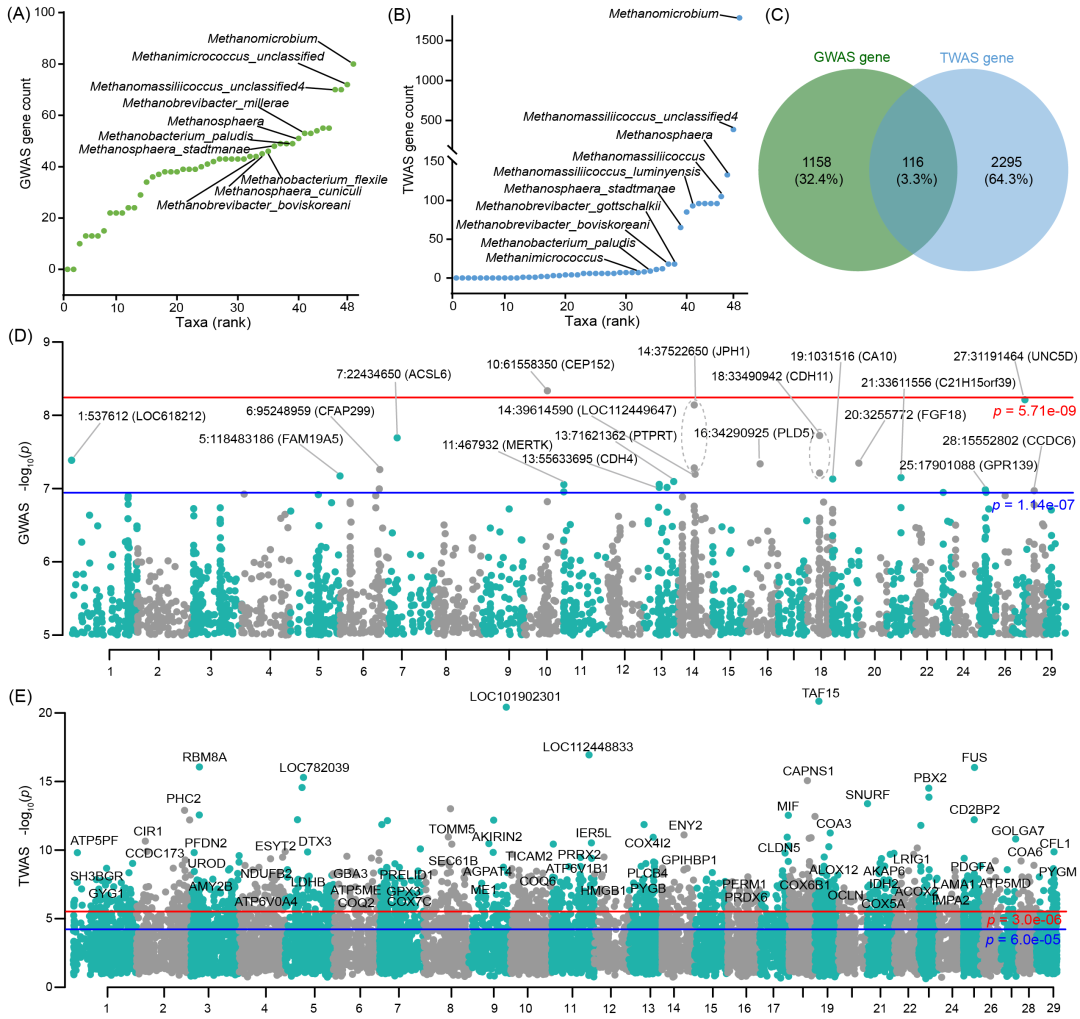
图4. 瘤胃古菌相关的宿主关联特征
(A)48个瘤胃古菌类群的GWAS注释邻近基因排序图;(B)48个瘤胃古菌类群的TWAS基因排序图;(C)GWAS和TWAS方法鉴定到的共享基因;(D)瘤胃古菌类群GWAS结果整合曼哈顿图;(E)瘤胃古菌类群TWAS结果整合曼哈顿图。曼哈顿图显示了每个SNP和基因的最小p值。红色和蓝色线分别代表全基因组显著性和建议显著性阈值。
多重相关性分析挖掘甲烷生成中的候选基因和菌群
为了揭示宿主瘤胃基因、瘤胃微生物和瘤胃液挥发性脂肪酸(VFAs)在牛瘤胃甲烷生成中的作用,我们整合了一个三重相关关系网络(基因-菌群-VFAs)。首先,考虑到挥发性脂肪酸浓度可以间接反映瘤胃发酵模式和甲烷排放水平,我们测定了瘤胃挥发性脂肪酸(表S9),并分别将细菌和古菌分类群与VFAs进行相关性分析。结果表明,Bacteroidales_F082与乙酸和乙丙比(A/P)呈正相关,Prevotella与丙酸呈正相关,Clostridia_UCG-014与总VFAs呈正相关(图5A)。值得注意的是,对于古菌,我们发现甲烷短杆菌属(Methanobrevibacter)的4种产甲烷菌(Methanobrevibacter_boviskoreani、Methanobrevibacter_millerae、Methanobrevibacter_thaueri和Methanobrevibacter_gottschalkii)表现出一致的与乙酸和乙丙比呈正相关,但与丙酸呈负相关(图5B)。基于此,我们以这4种产甲烷菌构建了细菌-古菌互作网络,筛选到36种与它们相关的瘤胃细菌(|r| > 0.2)(图S10)。为了进一步探索与甲烷生成调控相关的宿主瘤胃表达基因,我们将上述40个菌群TWAS显著关联基因合并,并选择了252个富集在代谢通路中的基因(图S11)。随后,我们对这些宿主瘤胃基因与瘤胃微生物菌群进行了相关性分析。基于上述结果,我们建立了宿主瘤胃基因、瘤胃微生物及挥发性脂肪酸与产甲烷古菌之间的综合关系网络(图5C和表S10、S11)。

图5. 瘤胃基因-菌群-挥发性脂肪酸之间的多重相关性网络
(A)瘤胃细菌分类群与挥发性脂肪酸之间的的相关性热图;(B)瘤胃古菌类群与挥发性脂肪酸之间的的相关性热图;(C)宿主瘤胃基因、瘤胃微生物菌群及挥发性脂肪酸之间的多重相关性分析。图中只显示相关系数|r| > 0.2且矫正p值 < 0.05的标记。*表示p < 0.05,**表示p < 0.01。
甲烷的产生被认为是瘤胃中主要的氢(H2)汇,它是由产甲烷菌以H2和CO2为底物合成的,因此,瘤胃中氢代谢及其相关微生物可能与高低甲烷表型密切相关。从上述多重相关分析中,我们发现一些分类群(如Oscillospirales_UCG-011、Christensenellaceae_R-7_group和Oscillospiraceae_NK4A214_group等)与4种产甲烷菌、乙酸和乙丙比呈正相关,其可能是潜在的产乙酸菌(图S10、图5A和图6),这些微生物通过发酵产生乙酸和大量的氢气,而这些氢气可以被产甲烷古菌用来产生甲烷。另外一些分类群(如Prevotella、Prevotellaceae_UCG-003和Anaeroplasma等)与4种产甲烷菌呈负相关,但与丙酸呈正相关,这意味着这些微生物可能是潜在的产丙酸菌(图S10、图5A和图6)。前期研究报道称丙酸生成过程可能竞争性地消耗H2,降低H2在甲烷生成中的可用性。这与一项针对水牛的研究相一致,该研究发现普雷沃氏菌丰度高的个体具有较低的甲烷排放水平。此外,在宿主基因层面,我们发现在淀粉和糖原代谢通路中显著富集的基因(如AMY2B、PYGB、PYGM、GYG1和MGAT4A等)与产乙酸菌和4种产甲烷菌呈正相关(图5C和图6)。另外,我们还发现了多种参与线粒体电子呼吸链相关的蛋白酶基因,包括过氧化物酶(如GPX3和PRDX6)、辅酶(COQ2)、细胞色素c氧化酶(如COX5A和COX6B1)、ATP酶(如ATP5ME、ATP6V0A4、ATP5F1E和ATP6V1B1)等基因,这些基因与产丙酸菌呈正相关,但与4种产甲烷菌呈负相关(图5C和图6)。且所有这些正相关和负相关的候选基因在瘤胃组织和瘤胃上皮细胞类型中均有表达(图S12)。总之,我们阐明了宿主瘤胃基因、瘤胃微生物和挥发性脂肪酸在瘤胃甲烷生成过程中的潜在作用关系。这些候选微生物和宿主基因将为未来减缓反刍动物甲烷排放相关研究提供重要参考。
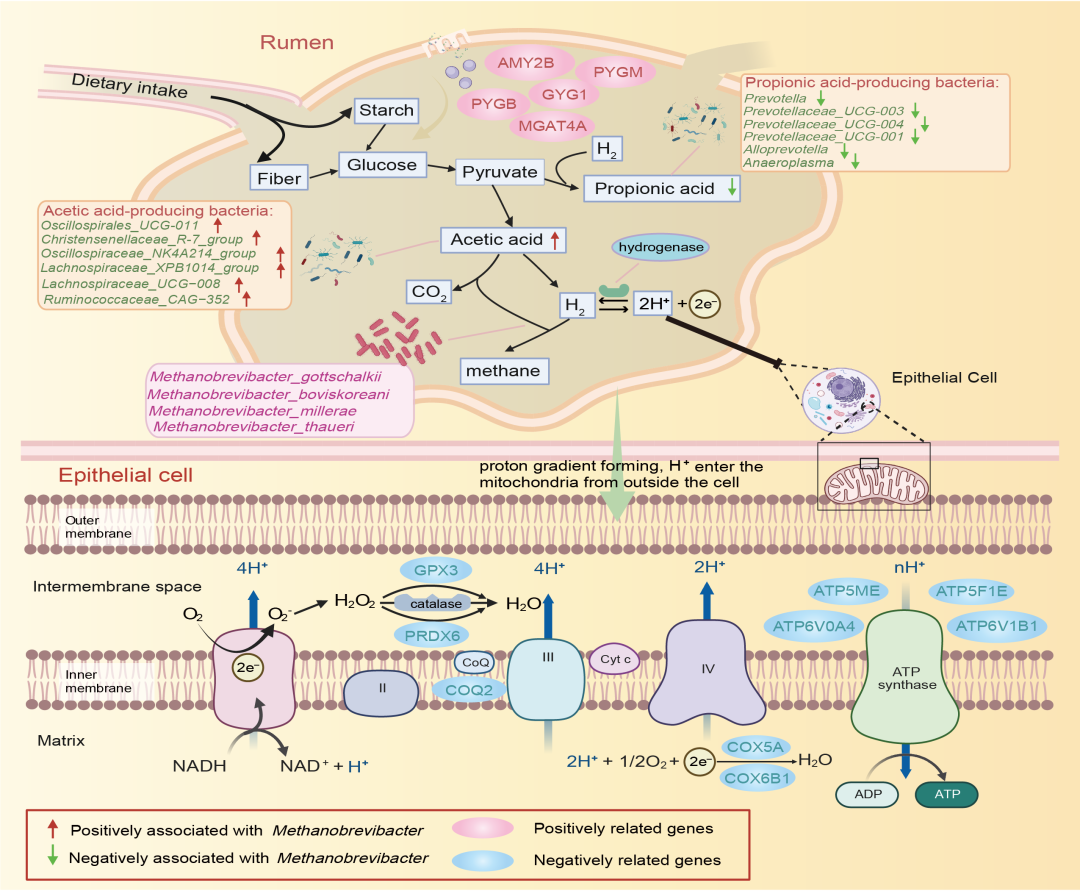
图6. 甲烷生成过程中假定的瘤胃碳水化合物和氢代谢途径概述
上半部分图说明了宿主瘤胃上皮淀粉和糖原代谢基因的高表达导致淀粉在瘤胃中更广泛地分解为葡萄糖的潜在过程,这削弱了利用淀粉的细菌生态位优势,增加了产乙酸盐微生物获得营养物质的机会,进而促进了瘤胃发酵向乙酸型模式的转变。乙酸型发酵导致产氢量增加,这反过来又增加了甲烷短杆菌属的丰度。下半部分图展示了宿主瘤胃上皮细胞线粒体氧化呼吸链中电子转移和氢离子运输的潜在过程。当瘤胃上皮氢离子转运和氧化磷酸化过程相关基因高表达时,宿主竞争性消耗氢离子,导致瘤胃中氢利用途径增加,从而降低了瘤胃中甲烷短杆菌属的丰度。图中候选微生物和宿主基因用不同颜色的箭头和椭圆形突出显示。
讨 论
反刍家畜的甲烷排放是农业温室气体的主要来源之一,在过去的几年里人们进行了大量的研究来寻找缓解策略。尽管越来越多的研究评估了宿主遗传、瘤胃微生物群和甲烷排放之间的关系,但甲烷排放是否可以通过牛对瘤胃微生物群的遗传效应来调节仍然存在争议。而且,以往的研究并没有阐明瘤胃效应器官中基因表达与微生物之间的关系。因此,牛瘤胃甲烷生成中的宿主-微生物组相互作用,在很大程度上仍然是未知的。为了更好地理解这一问题,我们首次整合了大规模群体瘤胃微生物的GWAS和TWAS分析,以研究宿主与产甲烷菌之间的调控关系。通过结合宿主瘤胃基因、瘤胃微生物和VFAs与产甲烷古菌之间的多重相关性分析,我们确定了多个参与甲烷生成的候选瘤胃基因和微生物菌群。这些初步结果将为通过遗传和微生物管理策略调控甲烷排放提供重要指导。
遗传力估计有助于理解宿主遗传对表型变异的影响程度。在这项研究中,我们发现瘤胃核心微生物群的遗传力可能几乎是普遍的,大约70%的瘤胃分类群表现出显著的遗传力,这与最近在绵羊中的一项研究结果一致。先前的研究证实,CH4的产生受到宿主遗传的影响,并表明甲烷排放具有中等遗传力,宿主遗传能够解释19-33%的甲烷排放表型差异。本研究中,我们的遗传力估计结果显示,瘤胃古菌也具有显著的遗传力,平均遗传力为0.41,如Methanobrevibacter_gottschalkii(h2 = 0.47)、Methanobrevibacter_boviskoreani(h2 = 0.33)和Methanobrevibacter_millerae (h2 = 0.27)(图2E和2H),这为我们在不直接测量甲烷的情况下寻找调控产甲烷古菌丰度的宿主遗传标记提供了重要线索。然而,在GWAS分析中,甲烷短杆菌属的产甲烷菌没有检测到显著关联的SNPs存在。这一结论与前人关于宿主遗传对牛瘤胃微生物群影响的大规模队列研究结果一致。鉴于在全球多项研究中,尚未发现与甲烷排放和产甲烷古菌丰度相关的主效信号位点。因此,我们推测甲烷生产可能是一种由宿主微效多基因控制的复杂表型,仅通过GWAS方法可能无法找到有效的遗传标记。
由于本研究收集了配对的转录组和微生物组数据,这一限制可以通过使用TWAS来解决,它可以直接鉴定与复杂性状显著相关的基因。在我们的研究中,我们首次对瘤胃微生物进行了TWAS分析,共检测到28,260个显著的基因-微生物关联(p < 3×10-6),涉及210种微生物分类群和4652个非冗余基因。这些宿主瘤胃上皮基因为我们研究甲烷生成过程中宿主-微生物相互作用提供了前所未有的机会。因此,我们整合了宿主瘤胃基因、瘤胃微生物及挥发性脂肪酸与产甲烷菌之间的三重关系网络,以挖掘参与甲烷生成途径的候选基因和微生物。我们发现甲烷短杆菌属的4种氢营养型产甲烷古菌(Methanobrevibacter_gottschalkii、Methanobrevibacter_boviskoreani、Methanobrevibacter_millerae和Methanobrevibacter_thaueri)与乙酸和乙丙比呈正相关,与丙酸呈负相关。在氢营养型产甲烷途径中,产甲烷菌通常将细菌发酵产物H2和CO2作为底物生成甲烷。因此,在某种程度上,一种很有前景的策略是调控底物H2的供应,以减少甲烷的产生。在我们的研究中,我们发现一些分类群(如Oscillospirales_UCG-011、Christensenellaceae_R-7_group和Oscillospiraceae_NK4A214_group等)与4种产甲烷菌、乙酸和乙酸丙比呈正相关,其可能是潜在的产乙酸菌。这些乙酸型发酵微生物产生大量的氢气,随后可以被产甲烷古菌用来生产甲烷。相反,另一些分类群(如Prevotella、Prevotellaceae_UCG-003和Anaeroplasma等)与4种产甲烷菌呈负相关,但与丙酸呈正相关(图5A和图6)。大量研究发现,普雷沃氏菌的丰度与甲烷排放呈负相关,表明普雷沃氏菌属的成员具有利用氢气生成丙酸而削弱甲烷生成的能力。
我们的TWAS和相关性分析结果显示,宿主瘤胃上皮基因与瘤胃微生物群之间存在显著相关关系。瘤胃上皮在VFA吸收、代谢和H+转运中起着重要作用。通常,瘤胃内的代谢氢([H])经过氢化酶对还原性辅因子的再氧化,将电子转移给H+形成H2(分子氢),这些H2被产甲烷菌用于产生甲烷。因此,我们推测宿主-微生物在能量代谢和甲烷产生中的相互作用主要是通过H+交换和运输发生。在本研究中,我们鉴定到252个与瘤胃细菌和产甲烷古菌相关的TWAS基因,这些基因在代谢通路中显著富集。其中,我们发现一些与产乙酸菌和4个产甲烷菌呈正相关的基因,其显著富集在淀粉和蔗糖代谢通路(如AMY2B、PYGB、PYGM、GYG1和MGAT4A等)。先前的一项研究发现,富含淀粉的饲料富含淀粉分解菌,以乳酸为中间体,通过丙烯酸酯途径促进丙酸的产生,减少产甲烷古菌对H2的可利用性,有助于维持健康的瘤胃。因此,我们推测宿主淀粉酶和糖原磷酸化酶相关基因可以竞争性地降解淀粉和糖原,从而降低了利用淀粉的细菌在瘤胃中的生态位优势,并增加了产乙酸盐的微生物获得营养的机会。这促进了瘤胃发酵由丙酸型向乙酸型转变,产生更多的氢气,供产甲烷菌用于甲烷生成。此外,瘤胃碳水化合物发酵还伴随着氢的代谢和运输,这些H2在微生物氢化酶的作用下通过H2→2H+ + 2e-发生可逆氧化反应。随后,这些H+通过质子梯度进入宿主瘤胃上皮细胞的线粒体电子呼吸链,参与能量代谢。我们的研究中还发现了大量的蛋白酶基因,如过氧化物酶(如GPX3和PRDX6)、辅酶(COQ2)、细胞色素c氧化酶(如COX5A和COX6B1)和ATP酶(如ATP5ME、ATP6V0A4、ATP5F1E和ATP6V1B1),这些基因与产丙酸菌呈正相关,与4种产甲烷菌呈负相关(图6)。这些基因可能通过介导呼吸链复合物中的电子转移和质子运输,参与线粒体呼吸链的ATP合成。呼吸链复合物I和III从分子氧(O2)中生成超氧化物(O2−)和过氧化氢(H2O2),这是线粒体中主要的活性氧(ROS)。谷胱甘肽过氧化物酶3基因(GPX3)和过氧化物酶6基因(PRDX6)是重要的过氧化物酶,它们催化过氧化氢降解为水,从而控制线粒体ROS水平,这一过程伴随着复合物I中NADH/NAD+的氧化还原反应和电子传递。细胞色素c氧化酶是线粒体呼吸链的末端酶,将氧气还原为水,从而促进电化学质子梯度的产生,驱动ATP合成。细胞色素c氧化酶亚基5A(COX5A)和细胞色素c氧化酶亚基6B1(COX6B1)是参与线粒体电子传递的细胞色素氧化酶基因,在线粒体功能障碍、氧化应激和细胞凋亡中起重要的保护作用。ATP酶基因(ATP5ME、ATP5F1E、ATP6V0A4和ATP6V1B1等)参与线粒体ATP合成,此过程主要利用H质子梯度从ADP和磷酸根离子合成ATP。上述结果表明,宿主瘤胃上皮细胞可能会竞争性地消耗H+参与线粒体呼吸链的能量代谢,导致用于甲烷生成的底物氢可用性降低,从而降低产甲烷菌的丰度。这一潜在机制与之前报道的结论一致,即高饲料效率的奶牛具有较低的产甲烷菌丰度。综上所述,这些研究结果表明,瘤胃甲烷生成过程中的宿主-微生物相互作用主要与氢代谢途径相关,未来需要更多的功能试验研究来阐明这些复杂的调控机制。
然而,本研究也存在一定的局限性。首先,微生物组数据是通过16S rRNA基因扩增子测序获得的,微生物基因和功能尚未阐明。其次,产甲烷菌丰度与甲烷排放量的关系尚不清楚,后续研究中,应测定部分个体的真实甲烷排放数据作为先验。此外,我们的研究使用了一个单一队列,这些研究结论是否普遍适用于反刍动物,需要在不同的牛群和更多其他反刍动物物种中进行验证。
结 论
本研究系统评估了宿主遗传变异和瘤胃基因表达对牛瘤胃微生物丰度变化的影响。我们发现,瘤胃效应器官的基因表达与瘤胃微生物丰度之间存在更直接的关系。研究结果强调TWAS是一种在基因表达水平上建立宿主和微生物关联很有前景的方法。通过结合多重关系网络(基因-菌群-挥发性脂肪酸),我们发现宿主-微生物在瘤胃甲烷生成中的相互作用主要涉及底物氢的代谢和转运。总之,这些发现为甲烷生成中宿主-微生物相互作用提供了新的见解,并为通过遗传调控和微生物管理策略减缓反刍动物的甲烷排放提供了有价值的指导。
方 法
动物样本采集
本研究共采集了574头成年雄性荷斯坦牛的样本。这些牛均为肥育公牛,年龄约2岁,体重相近,均来自中国宁夏地区的大型牧场。所有牛都是健康的,在定点屠宰场统一屠宰后收集瘤胃组织和瘤胃内容物样本。所有样本立即用液氮速冻,然后移入-80℃冰箱保存。
基因组测序和分析
采用标准的苯酚-氯仿法从瘤胃组织样本中提取宿主基因组DNA作为DNA文库制备材料。用DNBSEQ-T7平台对DNA文库进行测序,生成150 bp的双端reads。使用fastp软件过滤原始FASTQ序列,去除低质量reads。使用BWA软件将clean reads比对到牛参考基因组ARS-UCD1.2(GCF_002263795.1)上;进一步使用Picard v2.1去除重复序列。使用基因组分析工具包(GATK)中的“HaplotypeCaller”、“GenotypeGVCFs”和“selectvariant”程序进行SNP检出和基因分型。使用GATK的“Variant Filtration”模块过滤所有原始SNP,标准参数为“QD < 2.0, MQ < 40.0, FS > 60.0, SOR > 3.0, MQRankSum < - 12.5, ReadPosRankSum < - 8.0”。随后,使用PLINK对双等位基因变异进行更严格的质量控制,其过滤标准为:SNP检出率大于95%,样本检出率大于90%,次要等位基因频率大于5%。在这些步骤之后,总共获得了574个个体中的8,760,052个常染色体遗传变异,用于后续分析。
微生物扩增子测序和分析
按照制造商的说明,使用QIAamp Fast DNA Stool Mini Kit试剂盒从瘤胃内容物样品中提取微生物DNA。使用16S rRNA基因V3-V4区扩增细菌特异性序列,引物为341F:CCTAYGGGRBGCASCAG和806R:GGACTACNNGGGTATCTAAT。使用16S rRNA基因V6-V8区扩增古菌特异性序列,引物为915F:AGGAATTGGCGGGGGAGCAC和1386R:GCGGTGTGCAAGGAGC。文库构建成功后利用NovaSeq 6000平台对文库进行测序,细菌生成PE250双端序列,古菌生成PE300双端序列。原始双端Fastq序列用fastp软件进行质量过滤,并使用FLASH软件合并到tags。随后,使用QIIME2中的DADA2插件进行质量控制、去噪和嵌合体去除,得到高质量的有效序列。序列以99%的相似性聚类为扩增序列变体(ASV)。使用SILVA(v.138)数据库对ASVs进行物种分类学注释。采用QIIME2软件计算Alpha多样性指标和基于Bray-Curtis距离的Beta多样性。
转录组测序和分析
使用TRIzol试剂从瘤胃组织中提取总RNA。高质量的RNA样品构建测序文库,使用DNBSEQ-T7平台对文库进行测序,生成150 bp双端reads。原始FASTQ数据首先使用fastp软件进行质量过滤,然后使用HISAT2软件将clean reads比对到牛参考基因组ARS-UCD1.2。使用StringTie软件进行转录本组装,构建TPM标准化的基因表达矩阵。为了减少计算量和提高统计能力,我们保留至少在20%样本中TPM值大于0.1的基因,并去除异常个体(平均值±3sd)。最后,共有454个样本的16,674个基因被用于后续分析。
宿主遗传与瘤胃微生物关系研究
为了明确宿主遗传亲缘关系与微生物组成相似性之间的关系,我们首先使用GCTA软件计算了基于所有SNP基因型的遗传关系矩阵(GRM),然后使用自编R脚本将其转化为亲缘关系矩阵。微生物菌群相似性基于Bray-Curtis距离矩阵。采用Mantel检验,通过Spearman's秩相关rho评估宿主遗传亲缘关系与微生物组成相似性之间的相关性。
遗传力和表达力估计
采用GCTA软件中基于基因组的限制性最大似然法(GREML)评估不同水平分类单元的菌群遗传力,估算所有SNP解释的微生物差异。狭义遗传力用h2 =σg2/σp2表示,其中σg2和σp2分别为遗传方差和表型方差。类似地,“表达力”(e2)被用来估计所有基因解释的微生物方差。使用OSCA软件中的OREML模块来估计菌群表达力,由基因表达方差解释的微生物方差估计用e2 =σg2/σp2 表示,其中σg2和σp2分别为基因表达方差和表型方差。使用似然比检验计算遗传力和表达力估计值的p值,p值小于0.05认为具有显著的遗传力和表达力。
微生物全基因组关联研究(mbGWAS)
在我们的研究中,我们首先将出现率大于80%的分类群定义为数量性状表型(相对丰度),将出现在20%~80%个体中的分类群定义为二元性状表型(存在/缺失),而出现在少于20%个体中的分类群被丢弃。然后,我们使用GEMMA软件中的线性混合模型对分类群的相对丰度或存在/缺失性状进行mbGWAS分析。采用Bonferroni校正p值以确定显著关联位点,全基因组显著阈值设为5.71×10-9(0.05/8,760,052),建议性显著阈值设为1.14×10-7(1/8,760,052)。使用R中的qqman和CMplot包绘制曼哈顿图和Q-Q图。鉴定出的SNP使用ANNOVAR进行基因组注释。为了在假定区域中划分独立的关联信号,我们使用PLINK软件(参数:--indep-pairwise 1000 10 0.2)对SNP进行了连锁不平衡评估,r2大于0.2的SNP对被认为高度连锁。
微生物全转录组关联研究(mbTWAS)
对于mbTWAS,共选择了454个具有转录组和微生物组数据的配对样本。利用OSCA软件中EWAS程序对16,674个基因和317个分类群进行关联分析。具体为,第一步读取基因表达数据,并以二进制格式保存,参数为:osca --efile myprofile.txt --gene-expression --make-bod --out myprofile。第二步更新标签信息,参数为:osca --befile myprofile --update-opi annotated.opi。最后,使用线性回归模型将基因表达和菌群丰度表型关联,参数为:osca --befile myprofile --pheno my.phen --qcovar my.qcovar --linear --out my。使用Bonferroni法确定显著性阈值(PTWAS = 0.05/16,674 = 3×10-6)。
瘤胃基因cis-eQTL鉴定及共定位分析
我们对454个个体的瘤胃基因表达表型进行了顺式eQTL鉴定。我们保留了至少在20%样本中TPM值大于0.1的基因,并对基因表达量经log10归一化处理。然后使用FastQTL软件中的线性回归模型进行cis-eQTL鉴定。我们将基因转录起始位点上下游1 Mb的窗口定义为顺式eQTL,以检测基因表达与基因组遗传变异的关联。我们首先以permutation模型(参数:--permute 1000 10000)进行cis-eQTL鉴定,计算经验p值,并使用FDR方法对多重检验的β-近似p值和经验p值进行校正。我们将至少有一个显著cis-eQTL(FDR < 0.05)的基因鉴定为eGene。然后,我们应用nominal模型鉴定显著关联的eGene-eSNP对,我们将最接近FDR等于0.05的eGene的经验p值定义为全基因组最小p值阈值,我们将nominal模型p值低于最小基因水平阈值的SNP视为显著cis-eQTL。为了检测瘤胃基因表达和瘤胃微生物表型的共同变异,我们首先将15,301个显著GWAS位点(p < 1×10-5)与eQTL分析中鉴定的416,109个显著eSNPs取交集,得到770个共同位点。我们进一步使用Coloc软件进行共定位分析,将后验概率值(PP.H4)具有95%可信度的SNP.PP.H4被考虑为两性状可能共享的因果变异。
瘤胃内容物挥发性脂肪酸测定
测定挥发性脂肪酸(VFA)时,首先将瘤胃液在4℃下13000 × g离心10 min。将1 mL上清与0.2 mL 25%的偏磷酸混合,在4℃下10000 × g离心15 min。随后,取2 mL上清液与200 μL巴豆酸混合,经0.45 μm过滤器过滤至样品瓶中。采用气相色谱法测定VFAs浓度。
数据统计分析
使用R软件包(v.4.3.1)和GraphPad Prism 9进行数据统计分析。组间差异分析采用student's t检验进行比较,结果用平均值±SEM表示。采用R软件“psych”包中的“corr.test”函数,计算瘤胃基因、微生物类群和VFAs之间的spearman相关性,并采用Benjamini-Hochberg方法矫正p值。所有检验中p值小于0.05认为显著。
代码和数据可用性
本研究所使用的原始测序数据已存储在国家基因库生命大数据平台(CNGBdb)(https://db.cngb.org/cnsa/)中,登录号为CNP0003615和CNP0005690。主要数据和代码已上传至Github网站,网址为:https://github.com/WeiWang-NWAFU/Host-microbiome-iMeta2024。更详细的数据信息可联系通讯作者获取。补充材料(文本、图、表、中文翻译版本或视频)也可从线上(http://www.imeta.science/)获取。
引文格式:
Wei Wang, Zhenyu Wei, Zhuohui Li, Jianrong Ren, Yanliang Song, Jingyi Xu, Anguo Liu, et al. 2024. “Integrating genome- and transcriptome-wide association studies to uncover the host-microbiome interactions in bovine rumen methanogenesis.” iMeta 3: e234. https://doi.org/10.1002/imt2.234.
作者简介

王伟(第一作者)
● 西北农林科技大学动物科技学院在读博士研究生。
● 研究方向为宿主遗传调控奶牛瘤胃菌群,以第一作者在iMeta、International Journal of Molecular Sciences、Genes、Microbial Pathogenesis等期刊发表SCI论文5篇。

魏振宇(第一作者)
● 西北农林科技大学动物科技学院在读博士研究生。
● 研究方向为奶牛复杂性状与转录表型遗传调控靶点挖掘,以第一作者(含共同)在iMeta、GENE、Animals等期刊发表SCI论文5篇,以第一发明人授权国家发明专利2件。

李卓辉(第一作者)
● 硕士研究生,2024年6月毕业于西北农林科技大学动物科技学院。
● 研究方向为反刍动物瘤胃微生物功能解析。以共同第一作者在iMeta、International Journal of Molecular Sciences期刊发表论文。

王禹(通讯作者)
● 西北农林科技大学动物科技学院教授,博士生导师。
● 研究方向为利用系统生物学理论,整合多组学数据,以进化视角解析反刍动物复杂性状的遗传基础。主持国家重点研发计划青年科学家项目、中国博士后创新人才培养计划项目、国家自然科学基金面上项目、陕西省优秀青年基金项目等国家、省部级项目多项。在Science、Genome Research、Proceedings of the Royal Society B: Biological Sciences、Zoological research等高影响力期刊上发表研究论文。

武圣儒(通讯作者)
● 西北农林科技大学动物科技学院副教授,博士生导师。
● 研究方向为反刍动物胃肠道健康调控与碳水化合物高效利用的宿主-微生物互作机制。陕西省三秦英才引进青年人才、陕西省科协托举青年人才。主持国家自然科学基金青年项目、三秦英才引进青年人才项目等国家或省部级项目7项。以第一/通讯作者在Microbiome、iMeta、Journal of Animal Science and Biotechnology、Animal Nutrition、Journal of Dairy Science、npj Biofilms and microbiomes等期刊发表中科院Top期刊论文30多篇。任iMeta期刊执行副主编及多种国际期刊审稿人。

李福勇(通讯作者)
● 浙江大学第一类“百人计划”研究员,博士生导师,国家高层次青年引进人才。
● 主要研究方向为消化道功能微生物组与宿主的交互作用,结合多组学技术、分子微生物学技术及经典微生物学技术,阐释宿主与肠道微生物在营养和遗传层面的交互作用,取得了一系列创新性学术成果。在Microbiome、BMC Biology、Cell Host & Microbe、ISME、Pharmacological Research等权威学术期刊发表SCI论文30篇,现担任Microbiome期刊动物微生物领域副主编。
更多推荐
(▼ 点击跳转)
iMeta | 引用13000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期

3卷1期

2卷2期封底

2卷4期封底

3卷2期

3卷3期

3卷3期封底

3卷4期

3卷4期封底

1卷1期
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百千华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表所有领域高影响力的研究、方法和综述,重点关注微生物组、生物信息、大数据和多组学等。目标是发表前10%(IF > 20)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!发行后相继被Google Scholar、ESCI、PubMed、DOAJ、Scopus等数据库收录!2024年6月获得首个影响因子23.7,位列全球SCI期刊前千分之五(107/21848),微生物学科2/161,仅低于Nature Reviews,同学科研究类期刊全球第一,中国大陆11/514!
“iMetaOmics” 是“iMeta” 子刊,主编由中国科学院北京生命科学研究院赵方庆研究员和香港中文大学于君教授担任,是定位IF>10的高水平综合期刊,欢迎投稿!
iMeta主页:
http://www.imeta.science
姊妹刊iMetaOmics主页:
http://www.imeta.science/imetaomics/
出版社iMeta主页:
https://onlinelibrary.wiley.com/journal/2770596x
出版社iMetaOmics主页:
https://onlinelibrary.wiley.com/journal/29969514
iMeta投稿:
https://wiley.atyponrex.com/journal/IMT2
iMetaOmics投稿:
https://wiley.atyponrex.com/journal/IMO2
邮箱:
office@imeta.science

















 4138
4138

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









