



点击蓝字 关注我们
肠道菌群人源化驱动猪体内代谢和免疫转录组发生人源化转变

iMetaOmics主页:http://www.imeta.science/imetaomics/
研究论文
● 原文链接: https://onlinelibrary.wiley.com/doi/10.1002/imo2.70034
● DOI: https://doi.org/10.1002/imo2.70034
● 2025年6月24日,中国科学院动物研究所赵勇、赵方庆、海棠等在iMetaOmics在线发表了题为“Microbiota humanization drives human-like metabolic and immune transcriptomic shifts in pigs”的文章。
● 本研究使用人、猪和肠道菌群人源化猪的粪便宏基因组、转录组、代谢组和蛋白组数据,系统评估了肠道微生物人源化驱动猪体内代谢和免疫转录组发生人源化转变,在人源化猪体内实现了血清代谢组的人源化重塑,并揭示了免疫细胞亚群的转录变化。这些发现推进了我们对人类胃肠道微生物如何在猪模型中影响宿主代谢和免疫功能的理解。
● 第一作者:张照奇、徐亚男、庞琨
● 通讯作者:赵勇(y.zhao1@siat.ac.cn)、赵方庆(zhfq@ioz.ac.cn)、海棠(haitang@ioz.ac.cn)
● 合作作者:吴昌鸿、赵晨旭、雷桐、张佳玉
● 主要单位:中国科学院动物研究所膜生物学国家重点实验室、中国科学院深圳先进技术研究院合成生物学研究所、深圳理工大学合成生物学学院、北京大学人民医院风湿免疫科、中国科学院动物研究所、北京干细胞与再生医学研究院、中国科学院动物研究所北方大动物研究基地、中国科学院动物研究所干细胞与生殖生物学国家重点实验室
亮 点
● 通过将猪的肠道菌群人源化,成功建立了肠道菌群人源化猪模型;
● 在人源化猪体内实现了血清代谢组的人源化重塑;
● 人源化猪体内免疫细胞亚群的转录重编程揭示了其向人源化代谢和生物合成活性的转变。
摘 要
猪因其生理和解剖学上与人类的相似性,被视为临床异种移植的有前景候选者,同时也是生物医学研究中重要的大型动物模型。然而,物种间在肠道菌群、免疫功能和代谢方面的差异仍然是主要障碍。为此,我们通过将人粪便菌群移植到经抗生素处理的猪体内,建立了肠道菌群人源化(GMH)猪模型。我们采用宏基因组学、靶向代谢组学和单细胞转录组学(scRNA-seq)分析,系统评估了菌群组成、血清代谢物和免疫细胞谱的变化。宏基因组分析显示,GMH猪的肠道菌群组成转向人源化特征,表现为拟杆菌纲(Bacteroidia)富集和芽孢杆菌纲(Bacilli)耗竭。代谢组学分析表明,GMH猪的血清代谢物谱更接近人类。在检测到的423种血清代谢物中,136种在对照猪中低于人类的代谢物在GMH猪中表达上调,而79种在对照猪中升高的代谢物在移植后下降。值得注意的是,与色氨酸代谢、胆汁酸生物合成和脂肪酸代谢相关的通路在GMH猪中增强,而碳相关代谢和糖酵解通路减弱,表明其代谢表型有人源化趋势。整合微生物和代谢物数据,鉴定出分别与拟杆菌纲和芽孢杆菌纲相关的20种和33种代谢物。对外周血单个核细胞(PBMCs)的scRNA-seq分析表明,GMH猪中T细胞、单核细胞和B细胞亚群的转录谱和组成发生了重塑。这些发现证明,人类粪便菌群可以重塑猪的全身代谢和免疫结构,为研究宿主-菌群相互作用以及推进异种移植和菌群治疗的应用提供了一个强大的大型动物平台。
视频解读
Bilibili:https://www.bilibili.com/video/BV12o3jzBEK5/
Youtube:https://youtu.be/IHTcjhfPRLo
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/imetaomics/
全文解读
引 言
猪因在生理和解剖学上与人类相似而被认为是非常有前景的临床异种移植器官供体。除了与器官捐献相关外,猪也是用于人类疾病、药理学和移植临床前研究的宝贵非灵长类大型动物模型。最近的进展使得将基因工程猪的心脏和肾脏移植到脑死亡人类受体以及选定的临床病例中成为可能,证明了基因改造在克服强大免疫和生理障碍方面的潜力。尽管取得了这些进展,但仍需进一步优化以提高猪与人之间的免疫和生理相容性,特别是在细胞通讯、代谢和菌群等方面。
在过去的二十年里,人类肠道菌群逐渐被认为是调节人类健康和疾病易感性的关键决定因素。虽然啮齿动物模型,特别是无菌小鼠和大鼠,已被广泛用于研究人类肠道微生物组成和功能,但由于显著的物种间差异,其转化相关性有限。为了解决这个问题,我们通过将人类粪便样本移植到经抗生素处理的猪体内,开发了人源化肠道菌群的猪模型。该模型为临床前研究以及潜在的临床应用(包括异种移植)提供了一个有前景的替代方案。虽然无菌猪在以往的人源化研究中使用过,但饲养和无菌要求等实际限制限制了其可扩展性。因此,我们采用了先前报道的方法:受体猪先用广谱抗生素进行预处理以耗竭内源性肠道菌群,然后使用冷冻保存的人粪便进行粪便菌群移植(FMT)。尽管粪便样本在收集后立即在-80°C下储存,但口服给药途径可能损害了严格厌氧菌的活力。此外,缺乏已知菌环境可能导致物种间微生物竞争,这可能影响了定植效率。
鉴于猪具有快速生长特性以及对营养和代谢调节的易感性,因此它是研究宿主-菌群相互作用的一个有价值的系统。然而,比较研究表明,猪肠道宏基因组包含的预测基因数量少于人类肠道宏基因组,并且在分类学和功能类别上表现出更高的α多样性但更低的β多样性。使动物模型肠道菌群人源化的努力主要局限于小鼠和新生无菌仔猪。然而,猪肠道菌群人源化对宿主血清代谢物、血清生化指标、以及免疫细胞比例和转录谱的功能性后果,仍然知之甚少。在本研究中,我们通过将人类粪便菌群移植到经抗生素处理的幼龄猪体内,建立了肠道菌群人源化猪。我们使用靶向代谢组学和单细胞RNA测序(scRNA-seq)评估了菌群人源化对宿主血清代谢组和免疫细胞群的影响。我们的研究结果提供了关于人类肠道菌群如何影响猪代谢和免疫的新见解。这项工作为开发用于人类生物医学研究的肠道菌群人源化猪模型奠定了基础性的一步,并且潜在地,为生成用于临床异种移植的优化猪供体奠定了基础,但这需要在功能获益方面得到进一步验证。
结 果
在猪体内建立人源化肠道菌群
为了构建具有人源化肠道菌群的猪,我们首先根据志愿者筛选标准(表 S1)收集了四名健康供体的粪便。实验组和对照组的猪均在4周龄时断奶,并接受广谱抗生素处理,随后进行粪便菌群移植(FMT),详见表S2。实验组(以下称为 GMH 猪)在第7至17周和第18至27周期间接受供体粪便。所有动物在FMT之前,在第5-7周和第17-18周期间接受了抗生素治疗,以减少内源性菌群(图 1A)。 为了评估微生物人源化的程度,我们在第17周和第21周收集了对照猪和GMH猪的粪便样本,使用供体粪便样本作为人类参考,进行了16S rRNA测序。在第17周,两组显示出相似的菌群谱。然而,到第21周,在第二次抗生素疗程之后,GMH猪的粪便菌群显著转向人源化组成,与对照组出现差异(图 S1)。 为了实现整合的跨组学比较,我们在27周龄时从两组收集了血浆、外周血单核细胞(PBMCs)、肠道组织、肝脏、肌肉和粪便样本。这些样本进行了宏基因组测序、靶向代谢组学、定量蛋白质组学和scRNA-seq(图 1B 和表 S3)。与之前的报道一致,对照猪的肠道菌群比人类丰富得多。使用加权 UniFrac 距离分析供体、对照猪和GMH猪的宏基因组数据表明,GMH猪的菌群组成显著更接近人类供体,而非对照猪(表 S4),尽管两个猪组的总体微生物丰富度和β多样性仍然高于人类供体(图 2A 和 2B)。 在门水平上,弯曲杆菌门(Campylobacterota)和螺旋体门(Spirochaetota)在人类和猪之间观察到显著差异(p < 0.05,图 2C 和 2D)。此外,芽孢杆菌门(Bacillota)和拟杆菌门(Bacteroidota)的相对丰度在对照猪和GMH猪之间存在显著差异(p < 0.05,图 2E)。对这三个发生变化的细菌门的纲水平比较显示,只有GMH猪中的拟杆菌纲(Bacteroidia)和芽孢杆菌纲(Bacilli)接近人类水平,并与对照猪出现差异(图 2F 和 S2)。具体而言,与对照猪相比,GMH组显示出拟杆菌纲比例增加,杆菌纲比例显著降低,表明其微生物组成更接近人类(图 2F, S2 和 S3)。 总的来说,这些结果表明,抗生素预处理后的FMT可以有效人源化猪的肠道菌群,确立了GMH猪作为转化研究中有价值的大型动物模型。
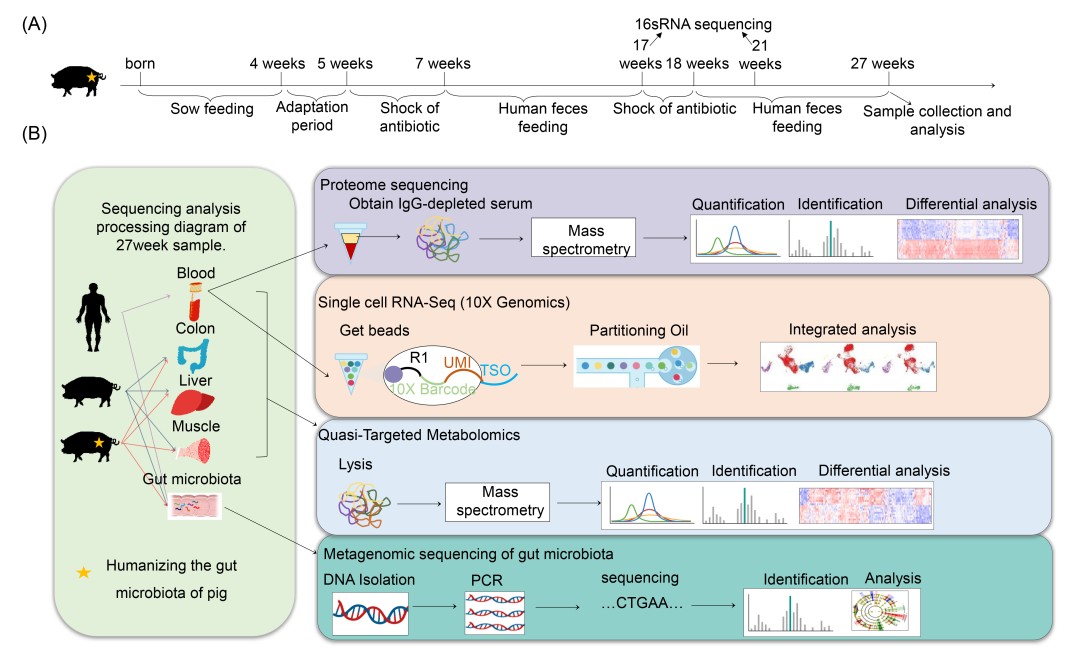
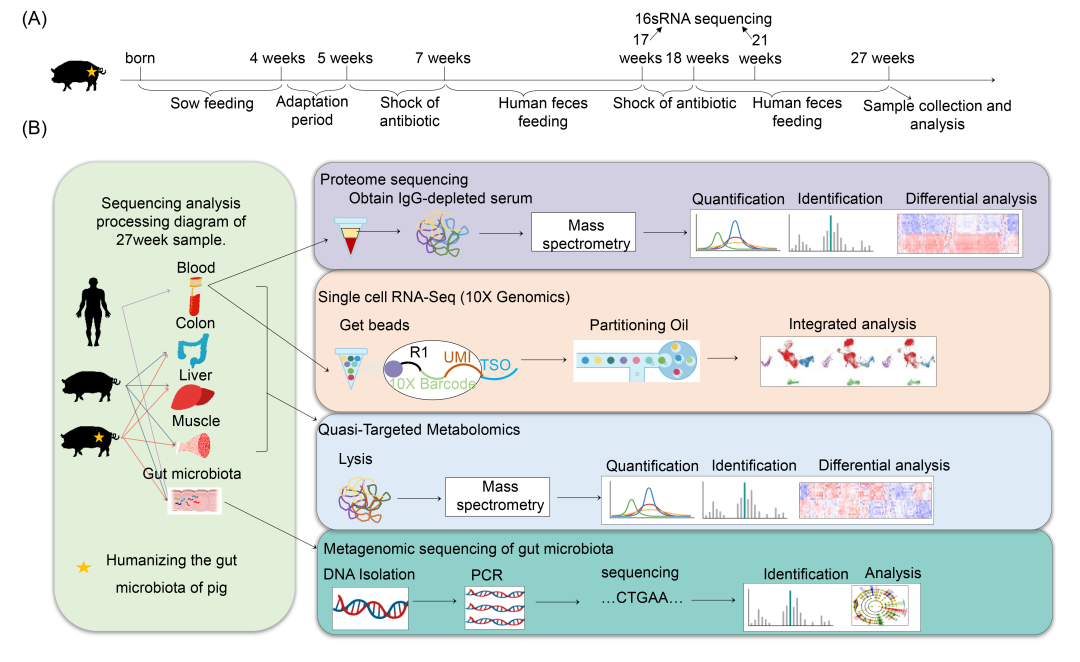
图 1. 人源菌群移植猪的构建
(A) 人源化猪的建立和分析流程。周数:不同处理下猪的周龄。(B) 本研究样本和分析流程示意图。我们收集了4个人的粪便并混合后饲喂猪。用于宏基因组测序的肠道菌群样本数量:4个人类粪便供体、6头对照猪和6头人类粪便移植猪。用于16sRNA测序的样本数量:4个人类粪便供体、6头对照猪和6头人类粪便移植猪。用于蛋白质组测序的样本数量:5个人类样本(4个粪便供体和1个额外样本)、6头猪和6头人类粪便移植猪。用于10X Genomics scRNA-Seq的样本数量:2个人类粪便供体、2头对照猪和2头人类粪便移植猪。用于血清靶向代谢组学的样本数量:5个人类、6头对照猪和6头人类粪便移植猪。用于结肠、肝脏和肌肉靶向代谢组学的样本数量:6头对照猪和6头人类粪便移植猪。
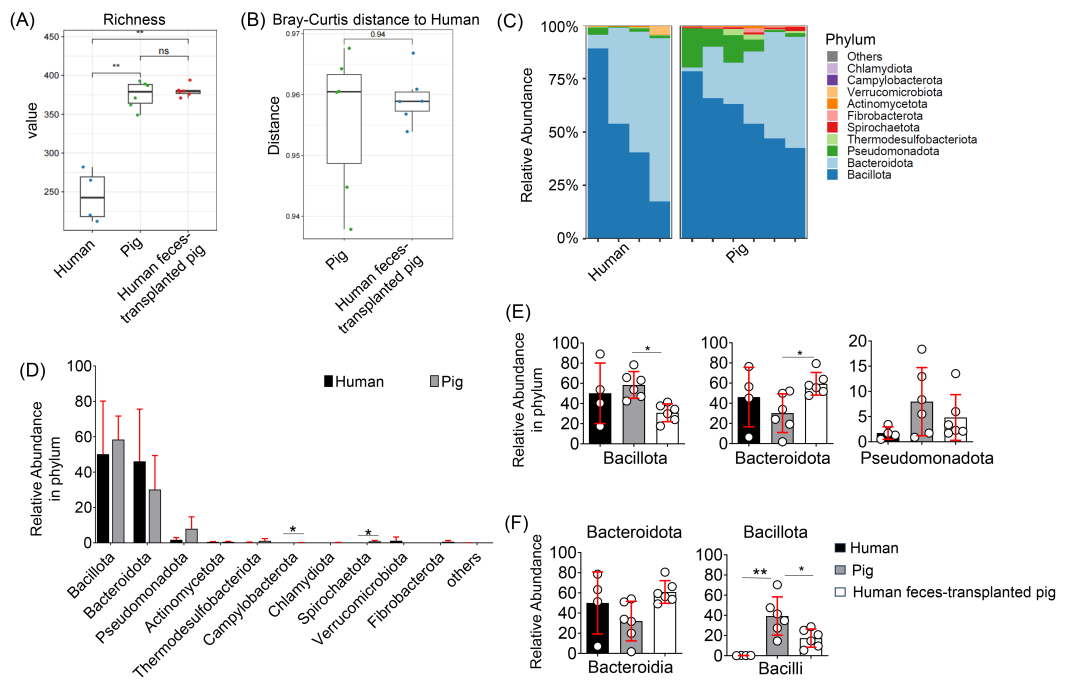
图 2. 人源化猪肠道微生物组的组成变化
(A) 肠道菌群丰富度分析统计图。(B) 对照猪和人源化猪与人类之间的Bray-Curtis距离分析。(C) 门水平样本组成。(D) 人类与猪之间门水平肠道菌群组成差异统计图。(E) 具有人源化趋势的门水平肠道菌群统计图。(F) 分别在拟杆菌门(Bacteroidota)和芽孢杆菌门(Bacillota)下具有人源化趋势的纲水平细菌群落。肠道菌群测序样本数量:4个人类粪便供体、6头对照猪和6头人源化猪。人类、猪和人源化猪样本中的数据表示为平均值±标准差(mean ± SD)。(*) p < 0.05, (**) p < 0.01,数据采用单因素方差分析(one-way ANOVA)检验,并进行Bonferroni校正。
人源化肠道菌群改变猪的血清代谢物谱
为了评估肠道菌群人源化的生理影响,我们在干预后每周测量了经抗生素处理的对照猪和 GMH 猪的体重和体温。两组之间在这两个参数上均未观察到显著差异(图 S4A 和 S4B)。同样,在第20周和第23周测量的血压和心率也未显示显著差异(图 S4C 和 S4D)。这些结果表明,人粪便移植并未损害猪的整体健康或生长。 已知菌群落通过产生广泛的代谢物影响宿主生理。为了确定人FMT是否可以将宿主全身代谢转向人源化状态,我们对27周龄时收集的对照猪(n = 6)和GMH猪(n = 6)的血清进行了靶向代谢组学分析(图 S5A)。为了进行跨物种比较,从供体独立收集了人类血清样本(n = 5)。在所有样本中,共鉴定出 423 种代谢物(图 S5B)。人和对照猪之间的比较分析揭示了显著的种间差异。对照猪显示出参与色氨酸、胆汁酸、鞘磷脂和亚油酸代谢的代谢物水平较低(图 S6A),而与精氨酸-脯氨酸代谢、丙酮酸代谢、磷酸戊糖途径和其他碳相关通路相关的代谢物水平较高(图 S6B)。值得注意的是,人粪便菌群移植显著减少了这些差异,将猪的血清代谢组转向人源化谱。在对照猪相对于人升高的 189 种代谢物中,有 79 种(41.8%)在 GMH 猪中降低(图 3A 和 3B)。相反,在猪中低于人类的 164 种代谢物中,有 136 种(82.9%)在移植后增加(图 3A 和 3B)。通路富集分析表明,在对照猪中富集的丙酮酸、磷酸戊糖和碳相关代谢通路在 GMH 猪中显著减少(图 S7 和 S8A)。相比之下,在对照猪中表达不足的色氨酸、胆汁酸和脂肪酸代谢通路在 GMH 猪中上调,趋向于人类水平(图 S7 和 S8B)。 这些代谢变化得到了临床血液生化分析的进一步支持,该分析显示与对照猪相比,GMH 猪的葡萄糖浓度较低,甘油三酯(TG)浓度较高,这与观察到的代谢组学趋势一致(图 S9)。此外,组织代谢组学显示出方向性变化:在血清中观察到的大多数代谢物变化也反映在结肠、肝脏和肌肉组织中(图 3A, S10A 和 S10B)。在 GMH 猪中,上调的血清代谢物主要与肌肉中发现的一致,而下调的代谢物则与结肠中的更密切相关(图 S10C 和 S10D)。相比之下,全局血清蛋白质组学分析显示GMH组和对照组之间无显著差异(图 S11),表明观察到的代谢效应并未伴随循环蛋白水平的变化。 为了进一步理解菌群-代谢物的相互作用,我们整合了人类、对照猪和GMH猪的宏基因组和代谢组数据集。相关性分析鉴定出20种与拟杆菌纲丰度呈正相关的代谢物,以及33种与杆菌纲相关的代谢物(图 3C)。这些代谢物涵盖多个化学类别,包括有机酸、有机杂环化合物、氨基酸、磷脂、酚酰胺和脂肪酰基(图 3D)。鉴于与对照猪相比,GMH猪显示出拟杆菌纲水平升高和杆菌纲水平降低(图 2F),我们推断这些分类群促成了观察到的代谢变化。具体而言,赤藓糖内酯(erythronolactone)、3-羟基异戊酸(3-hydroxyisovalerate)和 2-哌啶酮(2-piperidinone)与这两个纲均相关(图 3E)。其他代谢物,如 Phe-Phe、甜菜碱(betaine)、乳酸(lactic acid)、D-乳酸(D-lactic acid)、环己基乙酸(cyclohexaneacetic acid)、甘氨酰-L-缬氨酸(glycyl-L-valine)、(+/-)-章鱼胺盐酸盐((+/-)-octopamine HCl)和 6-羟基褪黑素(6-hydroxymelatonin)主要与拟杆菌纲相关,而异丁酰肉碱(isobutyryl carnitine)、二氢鞘氨醇(sphinganine)、Dl-二氢鞘氨醇(Dl-dihydrosphingosine)、溶血磷脂酰胆碱 15:0(lysoPC 15:0)和丁酸赖氨酸(lysine butyrate)则与杆菌纲更密切相关(图 3E 和 3F)。 综上所述,这些结果表明人类粪便菌群移植驱动了GMH猪中猪血清代谢组的显著重塑。这些变化与肠道菌群组成的改变密切相关,并将全身代谢谱转向人源化状态,支持了微生物和代谢人源化猪模型的成功建立。
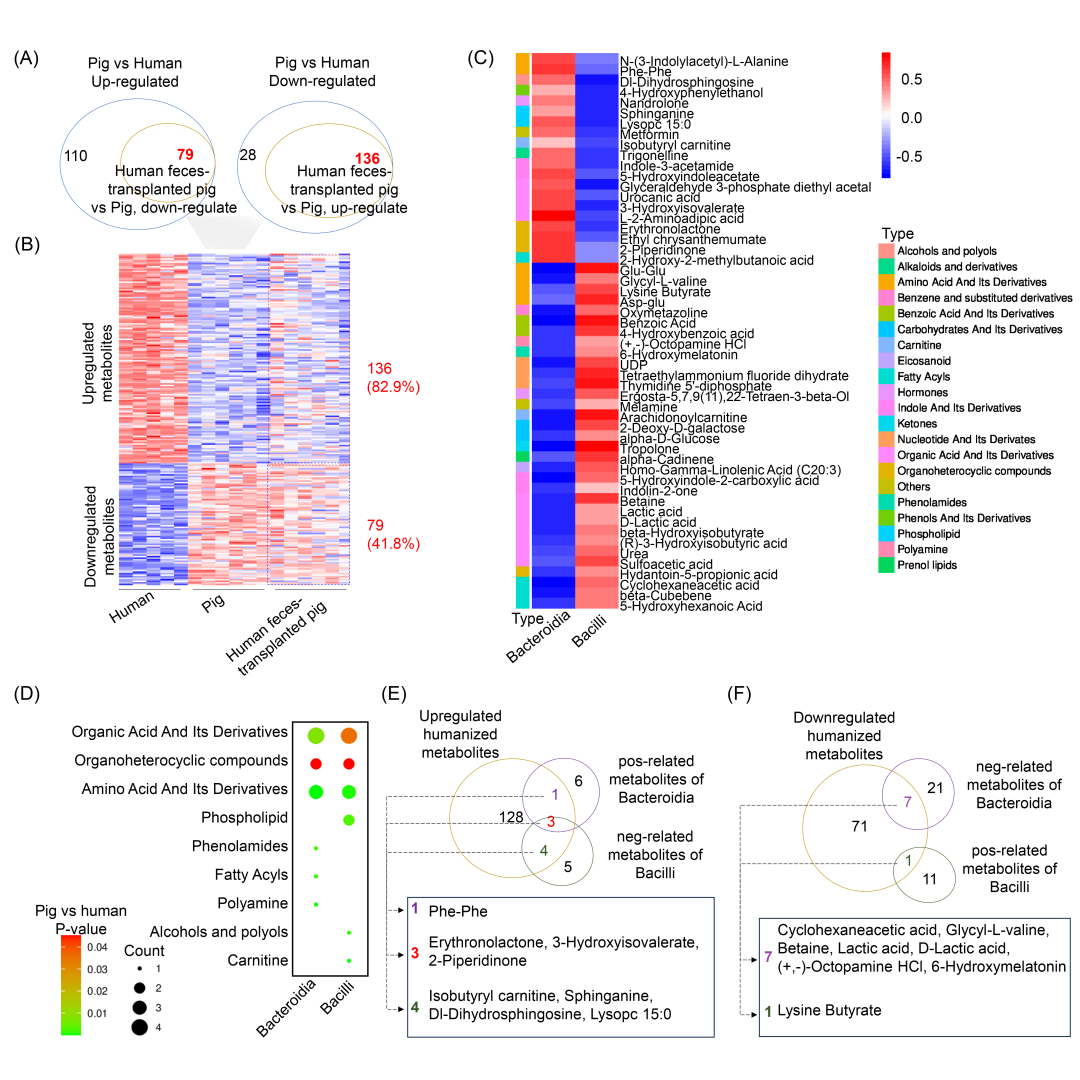
图 3. 肠道菌群对人源化猪血清代谢水平的影响
具有人源化趋势的血清代谢物的维恩图(A)和热图(B)。(C) 肠道菌群与代谢物含量相关性的热图。红色表示正相关,蓝色表示负相关。(D) 代谢物功能分析气泡图。受拟杆菌纲(Bacteroidia)和芽孢杆菌纲(Bacilli)调控的上调(E)和下调(F)人源化代谢物的维恩图。肠道菌群测序样本和血清代谢组测序样本数量:5个人类粪便供体、6头对照猪和6头人源化猪。
人、猪和GMH猪PBMCs的单细胞转录组图谱
为了在单细胞分辨率上比较跨物种的免疫细胞转录组谱,我们对来自人、对照猪和GMH猪的PBMCs进行了scRNA-seq。PBMCs从两名健康人类供体、两只对照猪(27周龄)和两只GMH猪(27周龄)中分离。使用10x Genomics 平台制备并测序单细胞文库(图 1A)。使用先前验证的策略,我们基于14,377个同源基因比对转录组。总共分析了来自人类的24,090个细胞,来自对照猪的20,402个细胞,以及来自GMH猪的15,837个细胞(表 S5 和图 S12)。三个数据集被整合并进行无监督聚类,初步鉴定出26个不同的细胞群(图 S13)。低表达PTPRC或富含血小板相关基因(簇 14、20、21 和 23)的簇被排除在下游分析之外(图 S14)。重新聚类后,我们解析出 31个转录上不同的免疫细胞群(图 S15)。注释以经典的人类免疫细胞标记物为指导,通过UMAP可视化了6个主要的免疫谱系,包括T细胞、自然杀伤(NK)细胞、细胞、CD14+ 单核细胞、CD16+单核细胞、树突状细胞(DCs)(图 4A, 4B 和表 S6)。跨物种谱系定义基因的保守表达支持了的分类及亚群的定义(图 4C 和 S16)。这些基因包括:T 细胞的CD8A、CD4、IL7R和CD3E;NK 细胞的NKG7和GNLY;B细胞的 CD19、CD79B和MS4A1;CD14单核细胞的 LYZ和CD14;CD16+单核细胞的 MS4A7和LYZ;以及DCs的FCER1A和CST3。亚群特异性转录调节因子和表面分子的表达水平,包括CD3E、CD4、CCR7、LEF1、TCF7、GNLY、NKG7、CD79A、LYZ和CST3,在人类和猪之间高度保守,验证了跨物种注释(图 4D)。 为了完善分类,我们应用了更新的人类-猪同源基因映射,并利用明确的人类 PBMC转录组参考来推断猪免疫亚群的分布。这个跨物种框架能够对各组间的免疫细胞比例进行比较量化(图 4E)。相对于人类,猪表现出更高比例的T细胞,但NK细胞和CD16+单核细胞的频率较低(图 4E)。值得注意的是,GMH猪显示出更接近人类的细胞组成,特别是在T细胞和CD14+单核细胞方面(图 4E),表明菌群驱动的免疫组成调节。这些结果建立了人、猪和GMH猪PBMCs的高分辨率单细胞图谱。
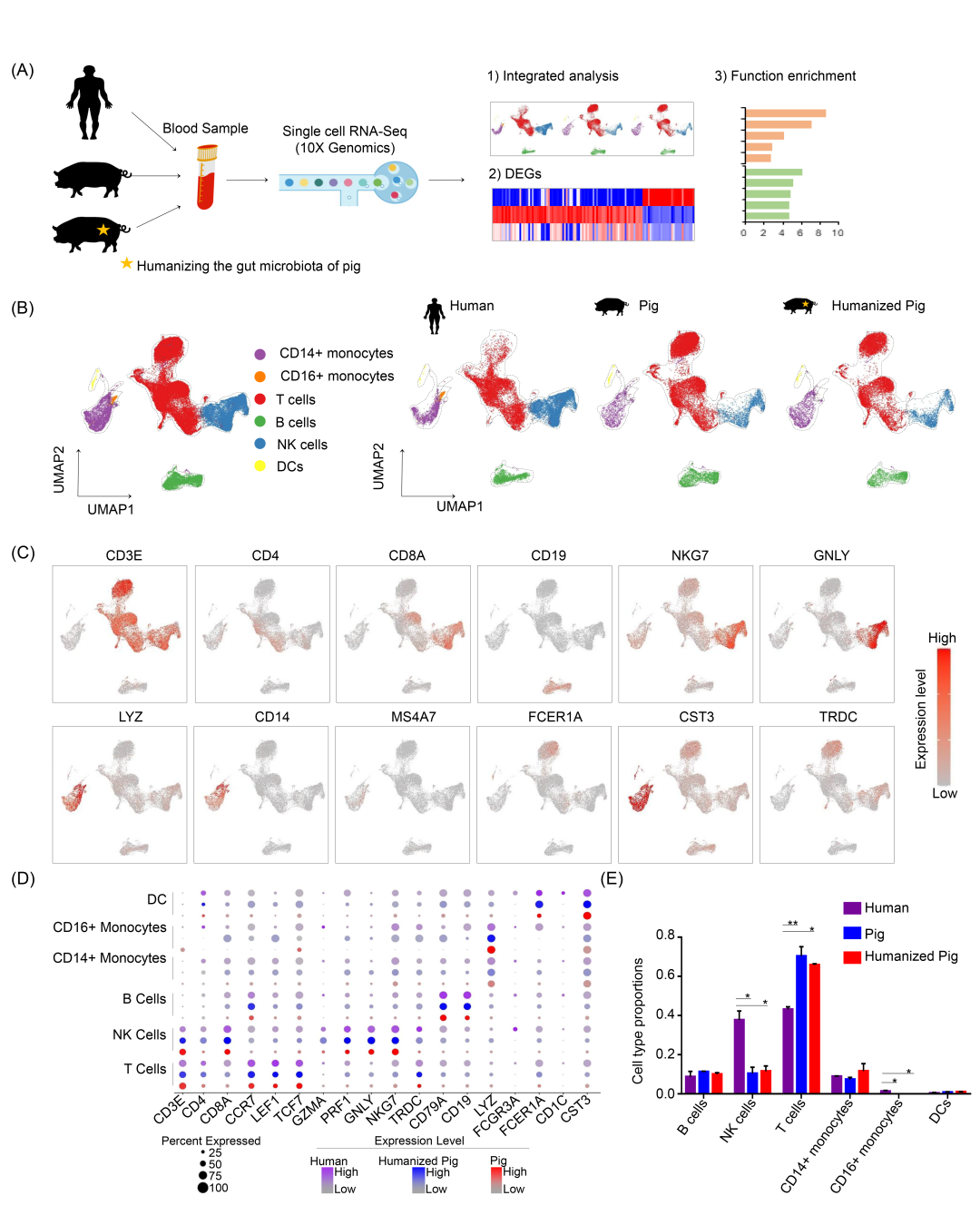
图 4. 基于PBMCs构建人-猪-人源化猪整合scRNA-seq图谱
(A) 本研究样本和分析流程示意图。(B) 人-猪-人源化猪整合聚类的UMAP可视化。以及所有细胞中分离的人类细胞、猪细胞和人源化猪细胞的UMAP可视化。每个点代表一个细胞,根据细胞类型着色。(C) UMAP特征图展示的免疫细胞标记物,UMAP图颜色表示所有细胞中基因表达水平。(D) 气泡图显示人、猪和人源化猪中标记物的表达水平。紫色代表人类中的表达水平,蓝色代表猪中的表达水平,红色代表人源化猪中的表达水平。(E) 人-猪-人源化猪细胞亚群比例统计图。用于scRNA测序的PBMCs样本数量:2个人类、2头猪和2头人类粪便移植猪。人类、猪和人源化猪样本中的数据表示为平均值±标准差。(*) p < 0.05, (**) p < 0.01,数据采用单因素方差分析检验,并进行Bonferroni校正。
人、猪和GMH猪中T细胞亚群的单细胞解析
T细胞是PBMCs的主要组成部分。为了解析跨物种的T细胞异质性,我们从整合的scRNA-seq数据集中提取了T细胞进行亚群聚类(图 S17)。基于已确立的人类T细胞基因特征,我们鉴定出两个主要的T细胞谱系:αβT细胞和 γδT细胞(图 5A 和 S18)。这些亚群的量化显示,αβT细胞在人类PBMCs中占主导地位,而猪T细胞中含有相对较高比例的γδT细胞(图 5B)。这种谱系分布在GMH猪中基本保持不变(图 5B)。接下来,我们根据基因表达特征进一步将γδT细胞细分为CD2-γδT细胞和CD2+γδT细胞(图 S19 和 S20A)。在GMH猪中,CD2- γδT细胞的相对丰度降低,表明该亚群可能受到菌群依赖性的调节(图 S20B)。功能富集分析显示,CD2- γδT细胞在与紧密连接、凋亡和吞噬体活性相关的通路中富集(图 S20C 和 S20D)。此外,该亚群表现出与细胞增殖和细胞周期相关基因(FOS、JUN、RHEX 和 SELL)的高表达,表明CD2- γδT细胞可能代表一个偏向于前体细胞的群体(图 S20E)。 为了详细研究肠道菌群人源化对αβT细胞状态的影响,我们从整合数据集中提取了αβT细胞进行高分辨率聚类(图 5A)。基于经典标记物,我们在跨物种中注释了6个不同的αβT细胞亚群:CD4+ naive T 细胞(CCR7、IL7R和CD4)、CD8+ naive T细胞(CCR7、IL7R和CD8A)、活化的CD4+ T细胞(CD4和S100A4)、GNLY+CD4+ T细胞(GNLY和CD4)、GNLY+CD8+ T细胞(GNLY和CD8A)和活化的CD8+ T细胞(CD8A和S100A4)(图 5C 和表 S7)。尽管这些αβT细胞的相对比例在GMH猪中未发生显著改变(图 5D),但转录组比较发现T细胞亚群的转录水平有人源化特征。具体而言,幼稚和活化的T细胞群在移植后表现出最大的转录变化,在CD4+ naive T细胞、CD8+ naive T细胞、活化的CD4+ T和活化的CD8+ T细胞中分别有687、771、800和735个基因显示出人源化表达模式。相比之下,终末分化的GNLY+ T细胞变化较小(图 5E)。 为了进一步探索功能变化,我们对在GMH猪T细胞中表达、且模式与人类细胞一致的基因进行了KEGG通路富集分析。在CD4+ naive T细胞和CD8+ naive T细胞中,上调的基因主要富集在生物合成和代谢通路中,包括剪接体组装、RNA 降解、氧化磷酸化、核糖体生物发生和产热。相反,与粘附连接、Th17细胞分化和白细胞迁移相关的通路在CD8+ naive T细胞中下调(图 5F)。类似地,在活化的CD4+和CD8+ T细胞中上调的基因富集在翻译和代谢过程,如核糖体、氧化磷酸化、RNA转运、产热和内质网(ER)中的蛋白质加工(图 5G)。GNLY+ CD8+ T 细胞也显示出核糖体和ER蛋白质加工通路的富集(图 5H)。 总之,这些发现表明人类粪便移植诱导了猪T细胞亚群的重塑。虽然总体亚群比例保持相对稳定,但幼稚和活化T细胞中的基因表达程序显著转向人源化模式,特别是在代谢和生物合成过程中。
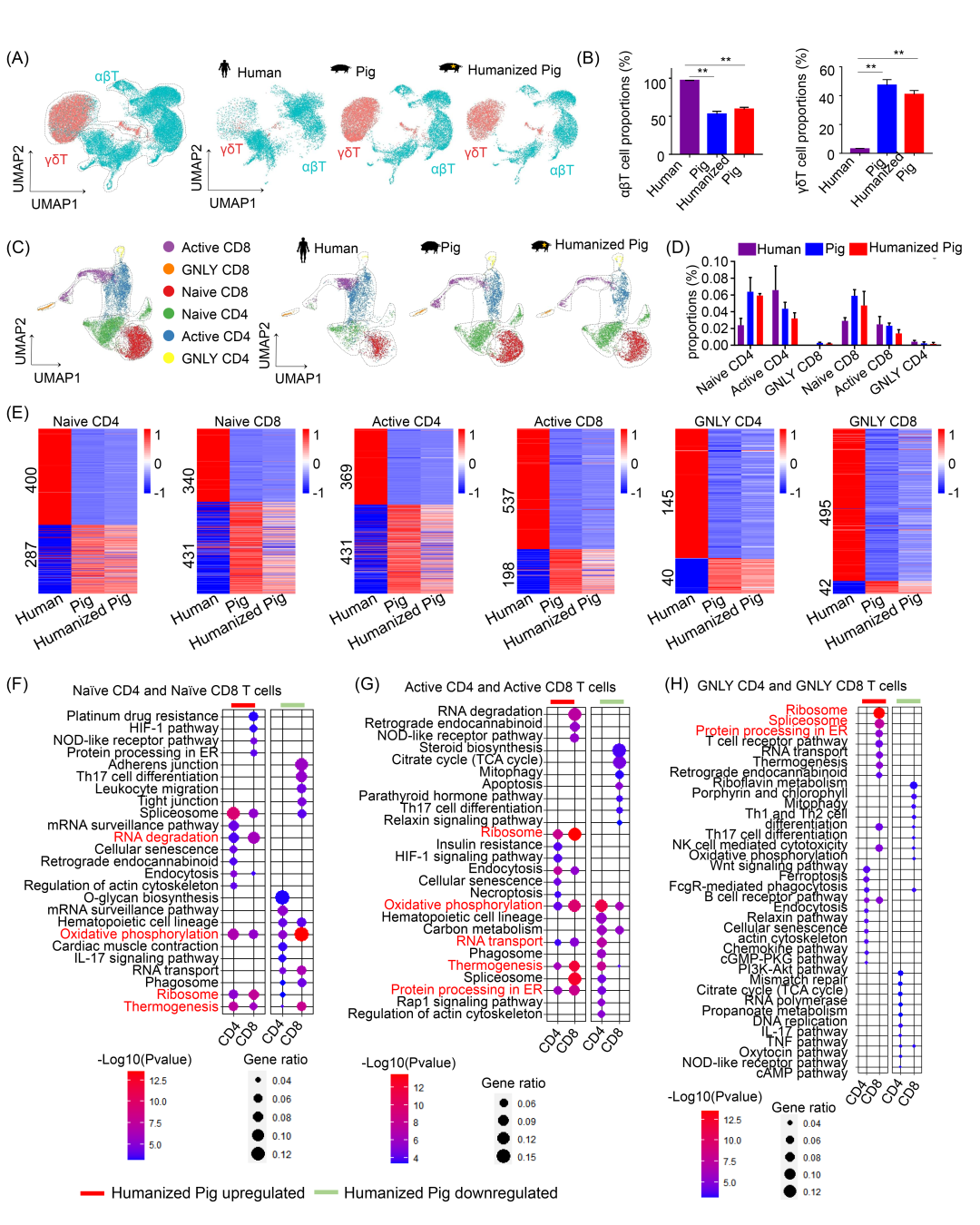
图 5. 从整合的人-猪-人源化猪scRNA-seq数据中解析和重新聚类T细胞
(A) 人-猪-人源化猪整合的αβ T细胞和γδ T细胞聚类的UMAP可视化图。粉色标记γδ T细胞,蓝色标记αβ T细胞。(B) 人类、猪和人源化猪中αβ T细胞和γδ T细胞的相对细胞比例(%)。(C) 人-猪-人源化猪整合的αβ T细胞聚类的UMAP可视化图。(D) 人类、猪和人源化猪中αβ T细胞亚群的相对细胞比例(%)。(E) 热图展示人类、猪和人源化猪中各类T细胞亚群中具有人源化趋势的基因。在幼稚T细胞(F)、活化T细胞(G)和GNLY+ T细胞(H)中,具有人源化趋势基因的KEGG富集分析。用于scRNA测序的PBMCs样本数量:2个人类、2头猪和2头人类粪便移植猪。人类、猪和人源化猪样本中的数据表示为平均值±标准差。(**) p < 0.01,数据采用单因素方差分析检验,并进行Bonferroni校正。
人、猪和GMH猪中髓系免疫细胞群的单细胞解析
为了研究人类、猪和菌群人源化猪中髓系细胞的组成和转录谱特征,我们从所有三组中提取并整合了PBMC来源的髓系细胞。无监督聚类基于经典的人类标记基因鉴定出4个主要群体,包括CD14+单核细胞、CD16+单核细胞、单核细胞来源的树突状细胞(monocyte-derived dendritic cells, mono-DCs)和浆细胞样树突状细胞(plasmacytoid dendritic cells, p-DCs)(图6A, 6B 和 S21)。定义每个亚群的高表达标记基因见图S22和表S8。其中,CD14+单核细胞在人类、猪和GMH猪中构成了主要的髓系亚群(图6C)。有趣的是,对照猪和GMH猪都表现出一个在转录水平上类似于人类CD16+单核细胞的独特簇,但这个群体本身CD16 FCGR3AFCGR3A的表达水平较低(图S21C),表明尽管具有共同的基因表达特征,但仍存在物种或条件特异性的差异。
为了系统地评估这些髓系亚群间的保守性,我们评估了在对照猪和GMH猪相应细胞类型中已确立的人类髓系标记基因的表达。总体而言,标记基因表达模式在物种和条件间高度保守。例如,CD14+单核细胞一致表达CD14、S100A8、C5AR1、S100A9和S100A12;mono-DCs表达PKIB、FCER1A、ITGB7、HLA-DOA和HLA-DQB1;而p-DCs表达FAM129C、ITM2C、CXCR3、PLAC8和VEGFB(图S21D)。
为了评估人类菌群定植对猪髓系细胞功能的影响,我们进行了对照猪、GMH猪和人类之间的差异基因表达分析。我们关注以下两类基因:在对照猪中相对于人类表达较高但在人源化猪中下调的基因;或在猪中相对于人类表达不足但在人源化后上调的基因(图6D)。在分析的4种细胞类型中,CD14+和CD16+单核细胞在人类粪便移植后表现出最大程度的转录重编程,分别有2505和921个差异表达基因(differentially expressed genes, DEGs)。相比之下,mono-DCs(504个DEGs)和p-DCs(193个DEGs)显示出较少的变化(图6D),表明单核细胞亚群对肠道菌群调节具有更高的响应性。
进一步对具有人源化特征的DEGs进行KEGG通路富集分析,我们发现,在CD14+和CD16+单核细胞中,粪便移植后上调的基因富集在与内质网(ER)中的蛋白质加工、产热、氧化磷酸化、内吞作用和MAPK信号传导相关的通路中(图6E),这与之前在T细胞中观察到的转录变化相似。在mono-DCs中,上调的基因主要富集在FcγR介导的吞噬作用、mRNA监视和长寿调节通路中,而下调的基因则与脂肪酸生物合成、磷酸肌醇代谢和昼夜节律导引相关(图6F)。在p-DCs中,上调的基因富集在诸如蛋白质输出、RNA降解、核糖体和内吞作用等通路中(图6F)。出乎意料的是,我们观察到在GMH猪的p-DCs中,突触相关通路显著下调。进一步的基因水平分析发现GNG2和GNG10(表S9)是这种富集的主要贡献者,它们是Ras信号通路的关键组成部分。这些发现表明,在人类菌群定植后,Ras介导的信号可能在猪的pDCs中被选择性地抑制。
综上所述,这些数据表明,人类肠道菌群的移植诱导了猪循环髓系细胞广泛且细胞类型特异性的转录重编程。单核细胞尤其表现出强烈的转录组学转变,趋向于人类样谱,表明肠道菌群具有调节全身性先天免疫功能的能力。
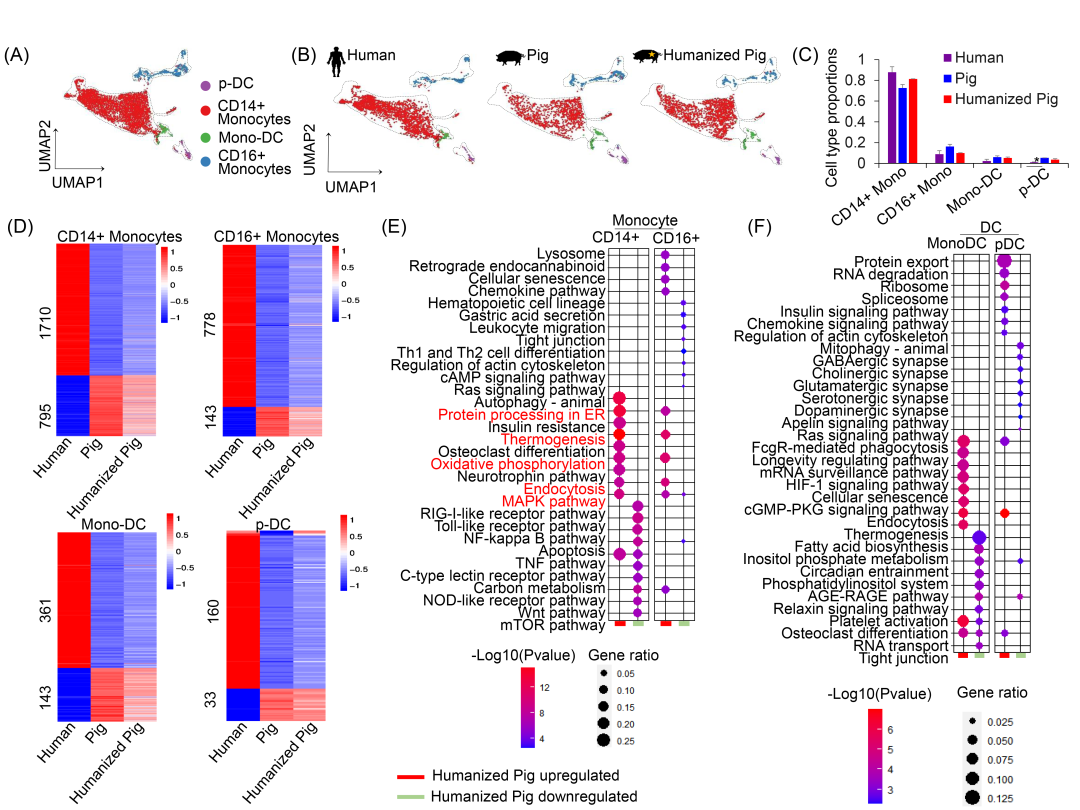
图 6. 从整合的人-猪-人源化猪scRNA-seq数据中解析髓系细胞的特征
(A) 人-猪-人源化猪整合的髓系细胞聚类的UMAP可视化。(B) 髓系细胞中分离的人类细胞、猪细胞和人源化猪细胞的UMAP可视化。不同颜色代表不同簇。(C) 人、猪和人源化猪中髓系细胞亚群的相对细胞比例(%)。(D) 热图展示人、猪和人源化猪中各类髓系细胞亚群中具有人源化趋势的基因。在单核细胞(E)和树突状细胞(DCs)(F)中,具有人源化趋势基因的KEGG富集分析。用于scRNA测序的PBMCs样本数量:2个人类、2头猪和2头人类粪便移植猪。人类、猪和人源化猪样本中的数据表示为平均值±标准差。
人类粪便菌群移植调节猪B细胞的组成和转录谱
与T细胞和单核细胞不同,B细胞在PBMCs中的总体比例在GMH猪、对照猪和人类之间保持相对稳定,未观察到显著差异(图4E)。为了研究人类粪便菌群移植是否诱导猪B细胞的表型和转录变化,我们从人、对照猪和GMH猪的整合scRNA-seq数据集中提取了B细胞。无监督聚类鉴定出5个B细胞亚群:幼稚B细胞(CD19+, CD79A+, CD2+, 和 CCR7+)、记忆B细胞(CD19+, CD79A+, CD2-, 和 CD27+)、CD24+ B细胞(CD19+, CD79A+, 和 CD24+)、CD5+ B细胞(CD19+, CD79A+, 和 CD5+)和浆细胞(CD19+, CD79A+, CD2-, 和 JCHAIN+),这些亚群是基于经典的人类B细胞标记基因注释的(图7A, 7B, 和 S23)。每个亚群的特征基因表达见图7C、S24和表S10。
在这些亚群中,CD24+ B细胞表现出最显著的组成变化。CD24是未成熟B细胞的一个特征明确的标记物,在B细胞成熟和信号传导中发挥作用。研究表明,猪中的CD24+ B细胞可能参与调节BCR信号传导和细胞增殖调控的基因。与之前的报道一致,CD24+ B细胞在对照猪中的频率高于人类。然而,在人类粪便菌群移植后,猪体内CD24+ B细胞的比例显著降低,接近在人类中观察到的水平(图7D)。这表明肠道菌群的组成可能影响B细胞的发育状态。
为了评估对粪便菌群移植的转录响应,我们以人类基因表达谱为参考,在人、对照猪和GMH猪的5个B细胞亚群中进行了差异基因表达分析。我们在幼稚B细胞、记忆B细胞、CD24+B细胞、CD5+ B细胞和浆细胞中鉴定出了一组在移植后表现出人源化基因(图7E-I)。对这些DEGs进行的GO富集分析进一步揭示了这种转变的功能意义。在对照猪中相对于人类表达较低、但在GMH猪中上调的基因,富集于与细胞周期进程、存活和B细胞发育相关的通路中。相反,在GMH猪中下调的基因则与对缺氧的反应、细胞增殖和细胞迁移相关。这些模式表明,肠道菌群人源化不仅改变了B细胞亚型的分布,还调节了与猪B细胞成熟和功能相关的核心生物学过程。
总之,这些结果表明,人类粪便菌群移植显著重塑了猪B细胞的组成和基因表达,促进了向更接近人类样免疫表型的转变。
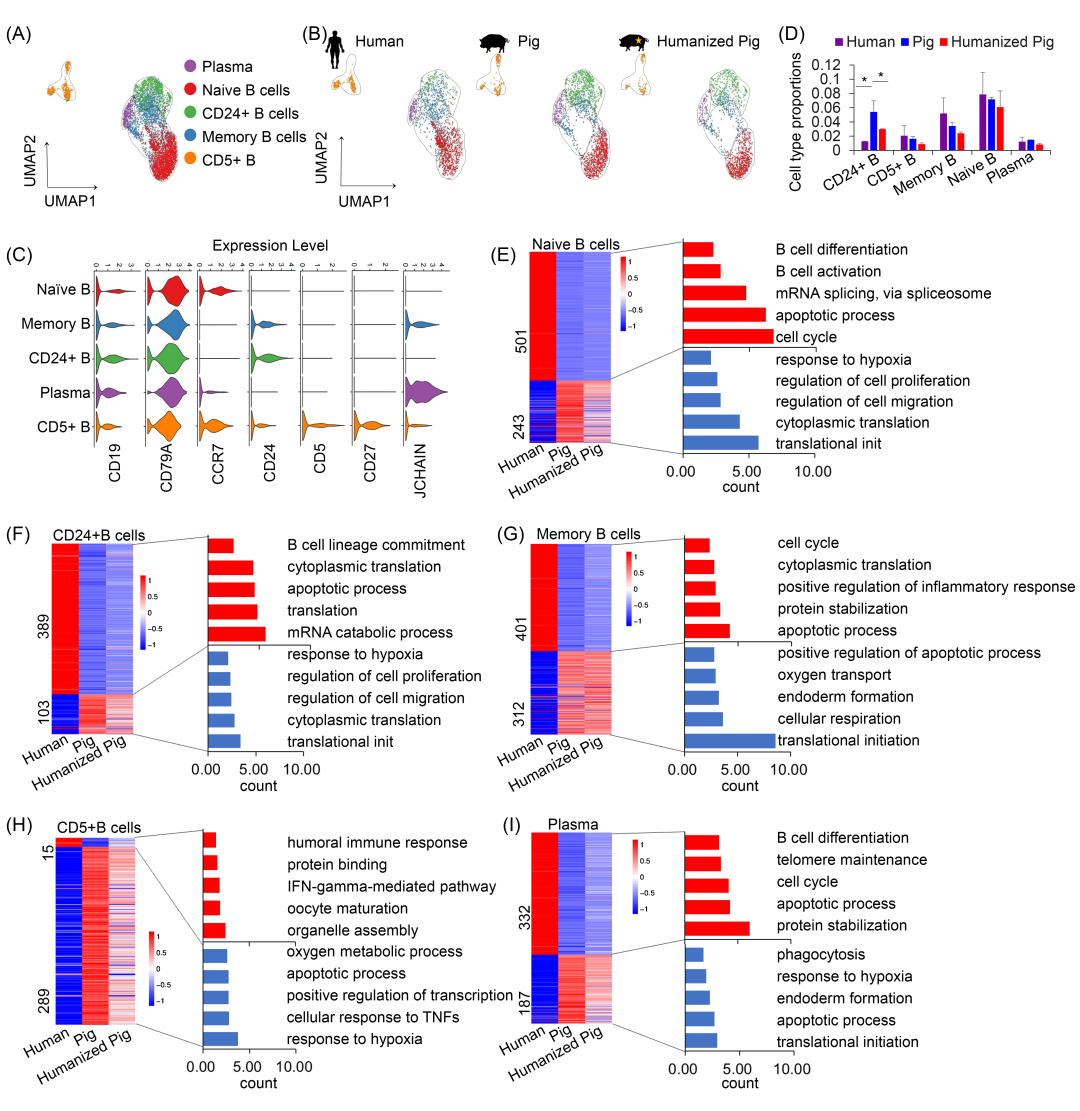
图 7. 从整合的人-猪-人源化猪scRNA-seq数据中解析和重新聚类B细胞
(A) 人-猪-人源化猪整合的B细胞聚类的UMAP可视化。(B) B细胞中分离的人类细胞、猪细胞和人源化猪细胞的UMAP可视化。不同颜色代表不同簇。(C) 小提琴图展示的特异性免疫细胞标记物,颜色标记不同簇。(D) 人类、猪和人源化猪中B细胞亚群的相对细胞比例(%)。在初始B细胞(E)、CD24+ B细胞(F)、记忆B细胞(G)、CD5+ B细胞(H)和浆细胞(I)中,具有人源化趋势基因的GO富集分析。用于scRNA测序的PBMCs样本数量:2个人类、2头猪和2头人类粪便移植猪。人类、猪和人源化猪样本中的数据表示为平均值±标准差。(*) p < 0.01,数据采用单因素方差分析检验,并进行Bonferroni校正。
讨 论
肠道菌群在调节人类健康和疾病易感性方面起着至关重要的作用。尽管啮齿动物模型,特别是无菌小鼠和大鼠,已被广泛用于研究人类肠道微生物的组成和功能,但由于显著的种间差异,其转化相关性有限。为了解决这个问题,我们通过将人类粪便样本移植到经抗生素处理的猪体内,开发了一种人源化肠道菌群的猪模型。该模型为临床前研究以及潜在的临床应用(包括异种移植)提供了一个有前景的替代方案。虽然无菌猪已在先前的人源化研究中使用过,但饲养和无菌要求等实际限制限制了它们的可扩展性。因此,我们采用了先前报道的方法:受体猪用广谱抗生素进行预处理以清除内源性肠道菌群,随后使用冷冻保存的人类粪便进行粪便菌群移植。尽管粪便样本在采集后立即于-80°C保存,但口服给药途径可能损害了专性厌氧菌的活性。此外,无菌环境的缺失可能导致种间微生物竞争,这可能影响了定植效率。
尽管存在这些局限性,我们的分析证明了受体猪体内成功的微生物定植和代谢重塑。宏基因组分析揭示了人源化猪中芽孢杆菌纲(Bacilli)和拟杆菌纲(Bacteroidia)的富集,这些菌纲在菌群人源化小鼠模型中也有关联 [27]。我们鉴定出136种在人源化猪中相对于对照猪显著上调、类似于人类谱的代谢物,以及79种下调至人类样水平的代谢物。相关性分析进一步揭示了20种与拟杆菌纲(Bacteroidia)呈正相关的代谢物,以及33种与芽孢杆菌纲(Bacilli)呈正相关的代谢物。值得注意的是,三种代谢物(赤藓糖醇内酯(Erythronolactone)、3-羟基异戊酸(3-Hydroxyisovalerate)、2-哌啶酮(2-Piperidinone))与拟杆菌纲和杆菌纲的丰度均呈强相关,强调了受人源化影响的潜在菌群-代谢物轴。
为了确定菌群人源化对宿主免疫的系统性影响,我们对来自人类、猪和GMH猪的PBMCs进行了整合单细胞RNA测序(scRNA-seq)。通过更新人-猪直系同源基因注释并应用经典的免疫细胞标记物,我们准确定义了主要的免疫亚群及其相对丰度。与人类相比,猪表现出更高比例的T细胞,特别是γδT细胞。粪便移植降低了猪体内T细胞的总丰度,并特别降低了CD2- γδT细胞的比例,使该谱图部分与人类谱图一致。尽管αβT细胞比例保持相对不变,但转录组分析显示,在GMH猪中,幼稚和效应T细胞群体(幼稚CD4+ T细胞、幼稚CD8+ T细胞、活化CD4+ T细胞、GNLY+CD4+ T细胞、GNLY+CD8+ T细胞和活化CD8+ T细胞)有部分基因表现出人源化趋势。粪便移植后猪T细胞中上调的基因主要富集在生物合成和代谢通路中,例如剪接体、RNA降解、氧化磷酸化、核糖体生物合成和产热。
在单核细胞群体中也观察到了类似的趋势。粪便菌群移植增加了CD14+单核细胞的丰度,该亚群在对照猪中比例不足。CD14+和CD16+单核细胞表现出最强的转录响应性,在肠道菌群人源化猪和对照猪之间分别有2505个和921个差异表达基因(DEGs)。这些基因富集在与内质网(ER)中的蛋白质加工、产热、氧化磷酸化、内吞作用和MAPK信号传导相关的通路中,进一步表明人类菌群移植后代谢激活增强。
B细胞亚群的变化也反映的肠道菌群移植的作用。我们从整合的人、猪和肠道菌群人源化猪scRNA-seq数据中鉴定出五个不同的B细胞亚群:幼稚B细胞(CD19+, CD79A+, CD2+ 和 CCR7+)、记忆B细胞(CD19+, CD79A+, CD2-, 和 CD27+)、CD24+ B细胞(CD19+, CD79A+, 和 CD24+)、CD5+ B细胞(CD19+, CD79A+, 和 CD5+)和浆细胞(CD19+, CD79A+, CD2-, 和 JCHAIN+)。亚群比例的对比分析显示,在对照猪中相对于人类比例较高的CD24+ B细胞,在GMH猪中显著减少,表现出趋近于人类B细胞谱图的转变。转录组分析进一步证明,初始B细胞和记忆B细胞对肠道菌群人源化特别敏感,在人源化猪与对照猪中分别有744个和713个DEGs。已知人类中的CD24+ B细胞在免疫介导的病理学中发挥调节功能并抑制过度的免疫反应。在猪中,CD24+ B细胞被表征为非典型记忆B细胞,可能由反复的BCR刺激产生,并与抗病毒体液免疫和炎症调节有关。我们先前的研究发现,猪CD24+ B细胞与人类T-bet+ B细胞共享转录组特征,后者参与对病原体的持续免疫反应。总之,这些发现支持了以下结论:肠道菌群人源化调节了CD24+ B细胞的比例和转录表型,使猪的B细胞格局更接近人类。
通过对血清代谢物、外周免疫细胞和肠道菌群的整合多组学分析,我们系统地描绘了人类粪便菌群移植诱导的分子和细胞改变。全面的转录组学和代谢组学分析揭示了基因调控网络的广泛重编程以及免疫细胞组成(包括T细胞、单核细胞、DC和B细胞)的显著变化。
使用在相同环境和饮食条件下维持的经抗生素处理的对照猪,提供了严格的实验对照,使我们能够将观察到的变化归因于肠道菌群人源化。虽然这些多组学数据集为肠道菌群的系统性影响提供了机制上的见解,但目前的发现本质上主要是相关性和计算性的。尽管我们纳入了选定的生化检测以支持观察到的趋势,但仍需进一步的体内功能验证,以建立微生物重组与宿主生理或免疫表型之间的因果关系。未来的研究应纳入靶向扰动研究,以确定人类来源的菌群分类群在这个大型动物模型中调节免疫功能和代谢稳态的具体作用。
结 论
通过将人类粪便菌群移植至经抗生素预处理的猪体内,我们成功建立了菌群人源化猪模型。该干预显著重塑了宿主的菌群落结构与血清代谢组特征,使代谢物谱趋近人类状态。单细胞转录组分析进一步揭示,菌群人源化可调控外周血免疫细胞亚群组成及基因表达谱,并在多类免疫细胞谱系中观察到代谢与生物合成特征的增强。这些发现共同证实,人类肠道菌群能够重编程大型动物宿主的全身代谢与免疫细胞功能。该菌群人源化猪模型为研究人类菌群-宿主互作、菌群相关疾病及微生态靶向疗法评估提供了转化相关平台,未来在异种移植研究和临床前安全性评估领域具备应用前景。
方 法
猪和人类样本收集
本研究使用的猪样本来自北方大动物研究基地。所有猪均按照中国科学院动物研究所《实验动物饲养和使用指南》饲养。本研究使用的人类样本来自健康成年个体。人类和猪的信息见表S3。所有程序均经中国科学院动物研究所生物医学研究伦理委员会批准(1OZ-IACUC-2021-008)。
抗生素处理
猪接受氨苄青霉素(Gold Biotechnology, St. Louis MO)60 mg-130 mg/天、万古霉素(Alfa Aesar, Ward Hill MA)15 mg/天、新霉素(EMD Millipore, Burlington MA)15 mg-30 mg/天和甲硝唑(Alfa Aesar)7.5 mg/kg的处理。
人类粪便的收集和保存方法
清晨收集新鲜粪便样本,置于无菌离心管或其他无菌容器中。将粪便挤入模具的小格中(约2 mL/格,称重约2 g),在液氮中快速冷冻3-5分钟,并于-80°C保存。
16S rRNA基因测序与分析
使用CTAB/SDS法提取所有微生物样本的总基因组DNA。通过1%琼脂糖凝胶电泳评估DNA浓度和纯度。使用区域特异
使用CTAB/SDS法提取所有微生物样本的总基因组DNA。通过1%琼脂糖凝胶电泳评估DNA浓度和纯度。使用区域特异性引物341F CCTAYGGGRBGCASCAGCCTAYGGGRBGCASCAG和806R GGACTACNNGGGTATCTAATGGACTACNNGGGTATCTAAT 扩增细菌16S rRNA基因的V3–V4高变区,每个引物均带有独特的条形码。使用Phusion High-Fidelity PCR Master Mix(New England Biolabs, USA)进行PCR扩增。将PCR产物与含有SYBR Green的1×上样缓冲液混合,并在2%琼脂糖凝胶上进行分析。选择在约400–450 bp处出现明显条带的样本进行下游处理。将来自不同样本的扩增子以等摩尔浓度混合,并使用Qiagen Gel Extraction Kit(Qiagen, Hilden, Germany)或GeneJET Genomic DNA Purification Kit(Thermo Scientific, Waltham, MA, USA)进行纯化。使用TruSeq DNA PCR-Free Sample Preparation Kit(Illumina, San Diego, CA, USA)制备测序文库,遵循制造商的方案,并为每个样本添加唯一的索引代码。使用Qubit® 2.0 Fluorometer(Thermo Scientific)和Agilent 2100 Bioanalyzer系统评估文库质量和浓度。在Illumina HiSeq平台上进行双端测序。 使用QIIME 2软件包(版本2020.2;https://docs.qiime2.org/2020.2/)处理原始序列数据。经过初步质量控制后,使用DADA2算法推断扩增子序列变体(Amplicon Sequence Variants, ASVs)。为减少噪音,排除在少于两个样本中检测到或总计数低于五的ASVs。通过将ASVs与SILVA参考数据库(版本138)进行比对,在99%序列一致性水平下聚类,使用QIIME 2预训练的进行分类器进行物种分类注释。生成ASVs的特征表,并在多个分类水平上计算每个分类单元的相对丰度,并将每个样本的总丰度归一化为100%。
引物341F CCTAYGGGRBGCASCAGCCTAYGGGRBGCASCAG和806R GGACTACNNGGGTATCTAATGGACTACNNGGGTATCTAAT 扩增细菌16S rRNA基因的V3–V4高变区,每个引物均带有独特的条形码。使用Phusion High-Fidelity PCR Master Mix(New England Biolabs, USA)进行PCR扩增。将PCR产物与含有SYBR Green的1×上样缓冲液混合,并在2%琼脂糖凝胶上进行分析。选择在约400–450 bp处出现明显条带的样本进行下游处理。将来自不同样本的扩增子以等摩尔浓度混合,并使用Qiagen Gel Extraction Kit(Qiagen, Hilden, Germany)或GeneJET Genomic DNA Purification Kit(Thermo Scientific, Waltham, MA, USA)进行纯化。使用TruSeq DNA PCR-Free Sample Preparation Kit(Illumina, San Diego, CA, USA)制备测序文库,遵循制造商的方案,并为每个样本添加唯一的索引代码。使用Qubit® 2.0 Fluorometer(Thermo Scientific)和Agilent 2100 Bioanalyzer系统评估文库质量和浓度。在Illumina HiSeq平台上进行双端测序。 使用QIIME 2软件包(版本2020.2;https://docs.qiime2.org/2020.2/)处理原始序列数据。经过初步质量控制后,使用DADA2算法推断扩增子序列变体(Amplicon Sequence Variants, ASVs)。为减少噪音,排除在少于两个样本中检测到或总计数低于五的ASVs。通过将ASVs与SILVA参考数据库(版本138)进行比对,在99%序列一致性水平下聚类,使用QIIME 2预训练的进行分类器进行物种分类注释。生成ASVs的特征表,并在多个分类水平上计算每个分类单元的相对丰度,并将每个样本的总丰度归一化为100%。
宏基因组测序与分析
使用天根DNA粪便提取试剂盒(TIANGEN Biotech Co., Ltd. China)按照制造商的方案提取RM粪便的总DNA。使用NanoDrop对提取的DNA进行定量。浓度 > 10 ng/μl 且 A260/A280 > 1.6 的DNA样本用于文库制备。最终文库的浓度 > 5 ng/μl,体积为50 μl。由诺禾致源科技有限公司(Novogene Co., Ltd.)进行Illumina测序。 对于宏基因组鸟枪法测序数据,使用trim-galore v0.5.0(q > 20)和cut-adapter v1.11去除低质量的原始双端读段和接头。使用Bowtie2 v2.1.0将过滤后的高质量读段与人类和猪基因组进行比对,以去除人和猪的污染。使用Kraken2 v2.1.3基于nr数据库对过滤后的宏基因组读段进行物种分类分析。确定每个样本中已鉴定分类单元的相对丰度,并分别在各个分类水平上归一化为100%的总丰度。
scRNA-seq文库构建
从猪的前腔静脉采集血液。通过Ficoll梯度离心分离外周血单个核细胞(PBMCs)。将单细胞悬浮于含有0.04% BSA的PBS中。每个通道加入约6,000个细胞,目标细胞数估计约为3,000个。捕获的细胞被裂解,释放的RNA在单个GEM中通过逆转录进行条形码标记。在S1000TM Touch Thermal Cycler(Bio-Rad)上进行逆转录:53°C 45分钟,随后85°C 5分钟,并在4°C下保存。生成cDNA后进行扩增,并使用Agilent 4200进行质量评估(由北京博奥晶典生物技术有限公司执行)。单细胞RNA-Seq文库制备:根据制造商的说明,使用Single Cell 3’ Library and Gel Bead Kit V3.1(10x Genomics, 1000075)构建单细胞RNA-seq文库。最终文库在Illumina Novaseq6000测序仪上进行测序,测序深度为每个细胞至少100,000个读段,采用双端150 bp(PE150)读长策略(由北京博奥晶典生物技术有限公司执行)。
scRNA-seq数据预处理
从10x Genomics网站获取Cell Ranger软件。使用Cell Ranger RNA mkfastq将原始测序数据转换为FASTQ格式。使用10x Genomics Cell Ranger RNA流程(版本1.2.0)完成scRNA-seq分析的比对。猪样本文库使用CellRanger-count(3.0.1)比对到索引的猪(Sus scrofa)Sscrofa10.2基因组。同时,我们使用我们先前文章中发表的人和猪的同源基因作为基因特征(14377个基因)。 我们将cellranger输出的数据矩阵读入R(v4.0.3),并使用Seurat(v4.0.1)进行处理。首先,我们设置参数min.cells = 3, min.features = 200来过滤细胞。过滤后获得的细胞数量为:human1 18,289, human2 18,569, Pig1 12,476, Pig2 12,609, Humanized Pig1 12,258, Humanized Pig2 12,466。然后计算每个细胞的线粒体相关基因("percent.mito")和核糖体相关基因("percent.ribo")的百分比。我们设置参数 nFeature_RNA > 200 & nFeature_RNA < 3000 & percent.mt < 20 & nCount_RNA > 500 & nCount_RNA < 15000 来过滤剩余的细胞。过滤后剩余的样本细胞数量见表S4。
富集分析
使用KOBAS软件(http://kobas.cbi.pku.edu.cn/)对基因进行GO富集、KEGG富集、Reactome富集和疾病富集。使用R包对结果进行可视化。从STRING数据库(https://string-db.org/)获取蛋白质-蛋白质相互作用网络,并使用Cytoscape(3.6.0)进行可视化。使用GSEA软件版本2.2.2.4进行基因集富集分析(GSEA)。
靶向代谢组学
收集人类、猪和人源化猪的血浆样本,送至诺禾致源科技有限公司进行处理和分析。将样本(100 μL)置于EP管中,用预冷的80%甲醇充分涡旋重悬。然后将样本在冰上孵育5分钟,并在4°C、15,000 g下离心20分钟。随后将样本转移至新的Eppendorf管中,在4°C、15,000 g下离心20分钟。最后,将上清液注入LC-MS/MS系统进行分析。使用KEGG数据库(http://www.genome.jp/kegg/)、HMDB数据库(http://www.hmdb.ca/)和Lipidmaps数据库(http://www.lipidmaps.org/)对代谢物进行注释。
数据统计分析
结果以平均值±标准差(means ± SD)表示。基于数据方差评估,使用非配对学生t检验(unpaired student’s t-test)检验两组间的差异。使用单因素方差分析(one-way ANOVA)检验数据,并使用Bonferroni校正进行多重比较。所有分析均使用GraphPad Prism软件(版本8)进行。* p < 0.05, **p < 0.01, ***p < 0.001。使用vegan R包计算丰富度(Richness)来评估Alpha多样性。使用vegan R包中的vegdist函数计算Bray–Curtis距离。基于相对丰度表,使用vegan R包中的adonis函数进行置换多元方差分析(Permutational Multivariate Analysis of Variance, PERMANOVA)来评估微生物群落水平的差异。
代码和数据可用性:
本研究中生成的人类PBMC RNA测序数据集可在基因表达综合数据库(Gene Expression Omnibus, GSE220297,有关GEO链接和引用的信息,请参阅:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE220297)中找到。本文报道的猪scRNA-seq数据已保存在中国科学院国家生物信息中心/北京基因组研究所(国家基因组科学数据中心)的OMIX数据库中(OMIX007789, https://ngdc.cncb.ac.cn/omix/release/OMIX007789; OMIX007790, https://ngdc.cncb.ac.cn/omix/release/OMIX007790; OMIX007791, https://ngdc.cncb.ac.cn/omix/release/OMIX007791; OMIX007792, https://ngdc.cncb.ac.cn/omix/release/OMIX007792)。 分析数据和脚本可在以下网址找到:https://github.com/zzqsherry/The-human-like-shifted-metabolomics-and-immune-cell-transcriptomics-in-gut-microbiota-humanized-pigs。补充材料(图、表、图文摘要、幻灯片、中文翻译版和更新材料)可在在线DOI或iMeta Science http://www.imeta.science/imetaomics/ 中找到。
引文格式:
Zhaoqi Zhang, Yanan Xu, Kun Pang, Changhong Wu, Chenxu Zhao, Tong Lei, Jiayu Zhang, et al. 2025. “Microbiota humanization drives human-like metabolic and immune transcriptomic shifts in pigs.” iMetaOmics 3: e70034. https://doi.org/10.1002/imo2.70034.
作者简介

张照奇(第一作者)
●中国科学院深圳先进技术研究院博士后,现北京大学人民医院,副研究员,硕士生导师。
● 主要从事自身免疫性疾病及相关微生物组、转录组数据挖掘等研究,主持国自然科学基金青年基金项目、深圳市医学研究专项人才提升项目、中国博士后基金第73批面上项目及中国科学院特别研究助理项目,以第一作者(含共同)在iMetaOmics、Science Advances、Cell Reports、The Journal of Immunology、iScience等多个期刊等期刊发表SCI论文13篇。

徐亚男(第一作者)
● 中国科学院动物研究所博士生,现复旦大学附属中山医院在站博士后。
● 研究方向为肿瘤免疫机制与免疫治疗,以第一作者(含共同)在iMetaOmics,Cellular & Molecular Immunology,Journal of Allergy and Clinical Immunology,EMBO Reports等期刊发表SCI论文9篇。

庞琨(第一作者)
● 中国科学院动物研究所在读博士研究生。
● 主要从事人体微生物组、单细胞及空间组学相关的数据挖掘及技术开发研究。目前以共同第一作者在Cell、Nature Genetics,Cell Reports、iMetaOmics等期刊发表SCI论文4篇。

赵勇(通讯作者)
● 深圳理工大学,合成生物学系,讲席教授,博士生导师。
● 研究方向:应用移植、自身免疫性疾病、肿瘤等动物模型,整合多组学及计算生物学,免疫学,分子生化及合成生物学等技术,进行免疫细胞功能极化的定量生物学分析,探讨免疫耐受及免疫应答调控的关键分子节点,筛选重要分子靶点并进行精准时空调控,创建可潜在应用于临床治疗的诱导或逆转免疫耐受治疗方案,开发新型免疫细胞药物及生物工程药物。国家杰出青年基金、新世纪百千万人才国家级人才、政府特殊津贴、中科院“百人计划”引进人才、“鹏城孔雀计划”特聘A档获得者。中国免疫学会移植免疫分会副主任委员,中国医药生物技术协会移植技术分会副主任委员。曾任中科院动物所副所长、膜生物学国家重点实验室主任、中国科学院大学存济医学院免疫教研室主任。长期从事移植免疫学研究,主要对移植免疫耐受诱导及其发生细胞分子机制和免疫细胞治疗进行研究,取得一系列重要研究成果。作为首席科学家主持科技部重点基础研究发展计划项目及国家自然基金重点项目等多项科研项目。在Nat Med、Nat Commun、Sci Adv、JEM、Blood、Cell Rep、EMBO Rep及Am J Transplant等发表SCI文章200余篇,中文核心期刊文章70余篇,获得发明专利授权1项。论文引用超12000次。分别获得爱思唯尔(Elsevier) 2021年和2022年“中国高被引学者(基础医学)”。主编《异种移植免疫学》及《移植免疫耐受》,参编《免疫学前沿进展》等5部书籍。

赵方庆(通讯作者)
● 中国科学院动物研究所,研究员,博士生导师。
● 研究方向为致力于建立高效的算法模型和实验技术,探索人体微生物与非编码RNA的结构组成与变化规律。基金委优青(2017)、北京杰青(2018)、国家杰青(2020)和中科院特聘研究员(2022)。现承担国家重点研发计划首席科学家、基金委杰青和重点项目、军委科技委基础加强计划首席科学家等项目。以第一/通讯作者在Cell、Gut、Nature Biotechnology、Nature Methods、Nature Genetics、Nature Cell Biology、Nature Computational Science和Nature Communications 等刊物上发表通讯作者论文100余篇,总引用次数超过1.5万次。现任中国生物信息学会基因组信息学分会主任、动物多样性保护与有害动物防控全国重点实验室主任。在Briefings in Bioinformatics、Science Bulletin、Science China Life Sciences、Genomics, Proteomics & Bioinformatics等国际学术刊物担任副主编或编委。

海棠(通讯作者)
● 中国科学院动物研究所高级工程师。
● 主要从事基于大动物的生物医学和农业育种应用,即通过大动物体细胞核移植、干细胞以及大规模基因筛查等技术平台建立异种器官构建、人类疾病大动物模型及家畜优良品种设计。相关研究获得了科技部重点研发计划,农业部转基因重大专项,国家自然科学基金,中国科学院先导专项以及中国科学院国际合作项目的资助,研究成果发表在Nature Biomedical Engineering, Cell Discovery, Cell Research, E life, Human Genetics, Protein&cells, Developmental Biology. 等杂志上。
更多推荐
(▼ 点击跳转)
iMeta | 引用16000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 兰大张东组:使用PhyloSuite进行分子系统发育及系统发育树的统计分析
iMeta | 唐海宝/张兴坦-用于比较基因组学分析的多功能分析套件JCVI
iMeta封面

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期

3卷1期

3卷2期

3卷3期

3卷4期

3卷5期

3卷6期

4卷1期

4卷2期

4卷3期
iMetaOmics封面

1卷1期

1卷2期

2卷1期

2卷2期
期刊简介
“iMeta” 是由威立、宏科学和本领域数千名华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表所有领域高影响力的研究、方法和综述,重点关注微生物组、生物信息、大数据和多组学等前沿交叉学科。目标是发表前10%(IF > 20)的高影响力论文。期刊特色包括中英双语图文、双语视频、可重复分析、图片打磨、60万用户的社交媒体宣传等。2022年2月正式创刊!相继被Google Scholar、PubMed、SCIE、ESI、DOAJ、Scopus等数据库收录!2025年6月影响因子33.2,中科院分区生物学1区Top,位列全球SCI期刊前千分之三(65/22249),微生物学科2/163,仅低于Nature Reviews,学科研究类期刊全球第一,中国大陆5/585!
“iMetaOmics” 是“iMeta” 子刊,主编由中国科学院北京生命科学研究院赵方庆研究员和香港中文大学于君教授担任,目标是成为影响因子大于10的高水平综合期刊,欢迎投稿!
"iMetaMed" 是“iMeta” 子刊,专注于医学、健康和生物技术领域,目标是成为影响因子大于15的医学综合类期刊,欢迎投稿!
iMeta主页:
http://www.imeta.science
姊妹刊iMetaOmics主页:
http://www.imeta.science/imetaomics/
出版社iMeta主页:
https://onlinelibrary.wiley.com/journal/2770596x
出版社iMetaOmics主页:
https://onlinelibrary.wiley.com/journal/29969514
出版社iMetaMed主页:
https://onlinelibrary.wiley.com/journal/3066988x
iMeta投稿:
https://wiley.atyponrex.com/journal/IMT2
iMetaOmics投稿:
https://wiley.atyponrex.com/journal/IMO2
iMetaMed投稿:
https://wiley.atyponrex.com/submission/dashboard?siteName=IMM3
邮箱:
office@imeta.science

















 1913
1913

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









