







1、打开VMD main上Extensions中的TkConsole这是VMD的控制命令区域。
2、围绕x轴旋转90:rotat x by 90 (或者rotat x to 90)
3、原子颜色设置+动力学键:
用fortran编写成lammps文件,可以识别原子类型,比如系统原子类型是1和2,点击VMD Main-Graphics-Colors-Categories选择Name-Names选择1或者2-Colors中选择颜色。
Graphics-Representations-Create Rep-Drawing Method-CPK设置Bond Radius为0,其余不变;Create Rep-Drawing Method-DynamicBonds设置Distance Cutoff为1.8,Bond Radius为0.2,Bond Resolution为6;
4、背景色改变:VMD Main-Graphics-Colors-Categories选择Display-Names选择Background-Colors中选择颜色。
5、前后原子隐藏:VMD main-Display-Display setting-Near Clip设置可使前面原子隐藏,Far Clip可使后面原子隐藏;Cue Mode中Cue Density可使原子更清晰。
6、坐标轴隐藏VMD main-Display-Axes-off
7、配合HyperSnap6截图:图像-剪裁可以截取任意尺度图像。
添加周期性水环境
package require solvate
solvate ubq.psf ubq.pdb -t 5 -o ubq wb
-t (override with any of the following)
-o (data will be written to output.psf/output.pdb)
添加离子中和多余电荷
autoionize -psf file.psf -pdb file.pdb [options]
可以直接使用VMD中extension>modeling>add ions的autoionize完成
TK console中选择全部原子,保存psf与pdb文件
参考:Autoionize Plugin
命令模式运行ubq.pgn
> vmd -dispdev text -e ubq.pgn
測量周期最大最小边界
set everyone [atomselect top all]
measure minmax $everyone
測量周期中心点
set everyone [atomselect top all]
measure center $everyone
測量质量中心
measure center $sel weight mass
位置移动
atomselect0 moveby {1 1 6} # 把所选原子向1,1,6向量方向和距离上移动
atomselect0 moveto { 3 6 5} # 把所选内容移动到3,6,5位置
原文:VMD的TK Console中的内置命令
设置resname/chain/resid
mol load pdb fileA.pdb
set sel [atomselect top "serial <=9"]
$sel set resname CD
$sel set chain X
$sel set resid 8
set all [atomselect top all]
$all writepdb fileB.pdb
refer the VMD Graphical Representations>Selections>Keyword for more values
top文件
IC A B C D [bond(AB)] [angle(ABC)] [dihedral(ABCD)] [angle(BCD)] [bond(CD)]
IC A B *C D [bond(AC)] [angle(BCA)] [improper(ABCD)] [angle(BCD)] [bond(CD)].
The * next to the C atom indicates that it is at the center of an improper angle definition. No * indicates that the 4 atoms do not have an improper topology.
Specifying IC’s is not necessary if you already have all the atoms in your PDB file. The entries are simply there
to have a way to create missing atoms from the positions of present ones.
参考:NAMD topology-tutorial
VMD键长单位
number in the “Value” field corresponds to the length of the bond in Ångstroms.
reference:Using VMD - An Introductory Tutorial
psfgen命令
pdbalias residue HOH TIP3 #aliasing residue HOH to TIP3
pdbalias atom TIP3 O OH2 #aliasing residue TIP3 atom O to OH2
vmd中tk命令打开文件
mol new filename.psf #打开一个文件
mol addfile filename.pdb #打开另一个文件,并叠加到第一个文件结构上
最适分子数
Presently, the maximum number of atoms one can expect to realistically compute with high level quantum chemistry is approximately 120 atoms
平衡态模拟
常见的模拟思路是,先在NVT下约束住你的溶质(剂)做限制性模拟,这是一个升温的过程,当温度达到你的设定后, 接着做NPT模拟,此过程将调整体系的压强进而使体系密度收敛。
经过一段时间的平衡模拟,在确定系统弛豫已经完全消除之后,就可以开始取数据了。如何判断体系达到平衡,简单的讲可以通过以 下几种方式,一,看能量(势能,动能和总能)是否收敛;二,看系统的压强,密度等等是否收敛;三看系统的RMSD是否达到你能接受的范围,等等。
升温/降温控制
# IF Heating
reassignFreq 2000
reassignTemp 100
reassignIncr 1
reassignHold 300
#从100K升温到300K,每2000步升高1K
# IF Cooling
reassignFreq 2000
reassignTemp 300
reassignIncr -1
reassignHold 100
固定分子
all atoms with a value of 1 (or a number different of 0) in a predetermined column will be fixed; atoms with a value of 0 in the same column will not be affected.
load pdb file into the psf file in vmd
in TKcon
set allatoms [atomselect top all]
$allatoms set beta 0
set fixedatom [atomselect top "resid 1 and name CA"]
$fixedatom set beta 1
$allatoms writepdb fileName.ref
拉动原子
to set which atom is to be pulled (SMD atom).uses the occupancy column of the pdb file to distinguish it
set allatoms [atomselect top all]
$allatoms set occupancy 0
set smdatom [atomselect top "resid 76 and name CA"]
$smdatom set occupancy 1
$allatoms writepdb fileName.ref
牵拉方向
set smdpos [lindex [$smdatom get {x y z}] 0]
set fixedpos [lindex [$fixedatom get {x y z}] 0]
vecnorm [vecsub $smdpos $fixedpos]
run a NAMD simulation
namd2 +p[procs] configfile > outfile
力场中二面角倒序正序区别
软件里的二面角有正负是得考虑4号原子在123号原子平面的哪一侧,举MS里二面角的来说,按顺序点1234共4个原子,二面角指的是123平面和234 平面的二面角,沿着23的轴看过去,1号原子在4号原子的逆时针方向(左手边),则取正值,反之取负值;绝对值仍在[0,180]。
原文摘录自:二面角和扭转角区别
Reversing the atoms in a proper dihedral does not have any mathematical or practical consequences. Conversely, the ordering of the atoms is very important for improper dihedrals.
原文摘录自:Parameters for a Protein-RNA covalent bond
力场参数图示
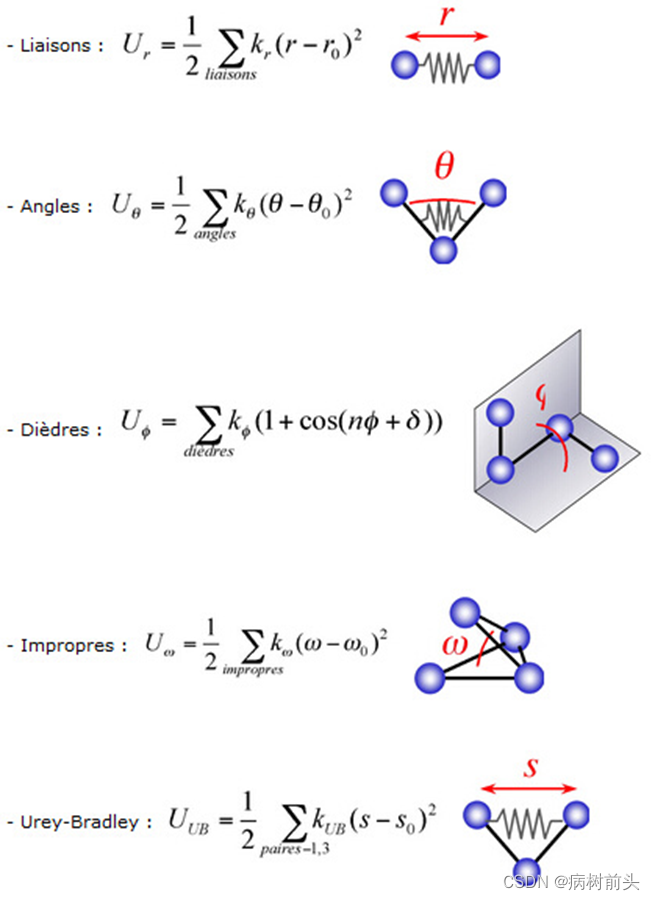
2014-06-18
图片来源:Brève introduction à la mécanique (MM) et à la dynamique moléculaire (DM)
improper
dihedral
By convention, the first atom of an improper
dihedral (type A-X-X-B or A-B-C-D) is usually the central atom. This had
been a general rule in the past.
2014-06-19
原文摘录自:CHARMM c32b2 parmfile.doc
拓扑文件AUTOGENERATE ANGLES DIHEDRAL
AUTOgenerate default options to be used when building a sturcture.AUTO ANGLes specifies that all possible angles and DIHEdral specifies that all possible dihedral angles be generated when building a structure. If these options are not included the angles and/or dihedrals must be listed explicitly in the topology file
2014-06-19
原文转自:CHARMM c38b1 rtop.doc
namd开始NPT计算过程报错Periodic cell has become too small for original patch grid解决方法
I have a different approach to fixing this problem. I believe that the underlying cause of this error is that the system is resizing too quickly (due to the system pressure being far from the specified pressure). To slow the resizing down I increase langevinPistonPeriod to 1000 and langevinPistonDecay to 500 for a short time when I start the NPT simulation, and then switch to the normal values of 100 and 50.
For example, in your case I would do one input file for 100 ps with a constraintscaling of 10, langevinPistonPeriod of 1000, and a langevinPistonDecay of 500. Then I would load the restart files into a new simulation and proceed as you were above. Also, by restarting the simulation you are forcing NAMD to re-assign the patches, which will help prevent this error.
2014-08-26
原文摘录自:Re: Periodic cell has become too small for original patch grid!
FATAL ERROR: Periodic cell has become too small for original patch grid!
问题出现在nvt计算完成后第一次进行npt的时候.通过在namd的邮件列表寻找,最后发现是周期性盒子设置的时候四舍五入数值,造成盒子在npt计算 过程中溶剂环境密度过小(盒子体积过大,而分子总数一定,最终水溶液密度过低)。解决方法,周期盒子大小不能直接四舍五入,要考虑到盒子体积与分子总数得 到溶液的密度是否恰当好处。改用该方法后,顺利计算,为提示错误。
2014-9-10
该内容原创
计算namd体系密度
首先计算体系的质量,然后根据namd输出的体积,两者相除即可。过程如下:
例如:
C 11
H 1596
O 790
N 2
上面表示的是每种原子的个数
则mass=(11*12+1596*1+790*16+2*14)/NA 单位为克。
NA为阿伏伽德罗常数:6.02214129(27)×1023
那么密度为mass/V
2014-08-26
原文摘录自:heating过程中水盒子形状发生变化
VMD计算SASA(solvent-accessible surface area )
控制台下用命令即可得出:
set all [atomselect top "all"]
set some [atomselect top "resid 1 to 5"]
measure sasa 1.4 $all -restrict $some -points sasapoints
foreach pt $sasapoints {
draw point $pt
}
以上可以在窗口中显示出1-5号氨基酸的溶剂可及表面,以point绘出。当然如果不需要浏览仅仅计算的话,只需要:
set all [atomselect top "all"]
set some [atomselect top "resid 1 to 5"]
measure sasa 1.4 $all -restrict $some
如果想看一下准不准可以算一下除了1-5号氨基酸的是多少
set someothers [atomselect top "not(resid 1 to 5)"]
measure sasa 1.4 $all -restrict $someothers
将两个值相加,看是否是
measure sasa 1.4 $all
得到的结果。
2014-9-10
原文摘录自:关于溶剂可及表面积(Solvent Accessible Surface)- Biocheming
try VMD timeline Tool to calculate the change in SASA over time.
Load the molecule and trajectory and then go to:
Extensions -> Analysis -> Timeline.
SASA is one of the many parameters you can calculate in the Timeline window.
2014-9-10
原文摘录自:How to calculate solvent accessible surface (SASA)?-Uttam Pal
旋转分子并保存文件
set sel [atomselect top "resid 10"]
set matrix [transaxis z 45]
$sel move $matrix
$sel writepdb out.pdb
20150405
原文参考:rotate a selection of a molecule and write rotated coordinates
linux系统ssh命令远程后台运行namd方法
nohup path/to/namd/namd2 +p 8 input.file.conf >out.file.log &
该方法确保ssh退出之后,远程主机仍然运行namd程序,+p后面的参数代表使用的cpu数量
以上命令在ubuntu 12.04/centos 5.5运行正常
20150407
原文参考:nohup Execute Commands After You Exit From a Shell Prompt
Getting ssh to execute a command in the background on target machine
tk console 更改切换硬盘位置
cd C:
cd D:
cd E:
更多示例
% pwd
c:/tcl
% cd d:
% pwd
d:/
% cd c:
c:/tcl
20150428
内容参考:tcl::cd command change the Windows Drive
1. 首先在调用vmd脚本的命令:vmd -dispdev text <psf> <dcd> -e script.tcl -args arguments
psf (NAMD)也在gromacs中对应gro, dcd对应trr。其实就是读入轨迹的过程,其实在脚本里在load进来这些分子也是可以的。-e后面即为这次操作的脚本 -args后面可以向脚本中传入参数,在脚本上可以通过lindex $argv n来调用,n对应为第几个参数,从0开始计数。
2. set variable [atomselect top protein]
variable即为变量名,后面可通过$variable调用。方括号里面表示选择top分子的protein部分。如果你load了很多分子,那么可以通过0,1,2....来调用。也可以在load的时候赋值给一个变量,这里就可以atomselect $variable ...了。
protein部分即为你想选择的部分,一般要加“”, 如果选择较复杂可通过括号,and,or来各部分
我接触到的一些选择:
protein and noh 蛋白质部分的重原子
backbone 主链
segid PROT 应该和protein一样
resid 残基序数 (1,2...)
resname 残基名称 (GLY...)
name 原子名称 (CA...)
3. mol load pdb "XXX.pdb"
在vmd中load一个pdb分子。同样可以放在方括号里赋值给一个变量。pdb也可以是gro啊,或其他top文件。
对于用amber力场模拟小分子的来说用parm7表示prmtop,rst7表示inpcrd文件。mol load parm7 "XXX.prmtop" rst7 "XXX.inpcrd"。
在load一个分子之后记得要用atomselect选择才能进行更多的操作。
4. molinfo top get numframes
molinfo可以得到分子的各种信息,上述语句就是用来得到MD模拟中轨迹文件的帧数,利用这个就可以构建循环语句对轨迹文件进行分析了。get后面加你想要得到信息的关键字。
5. measure命令
一个强大的命令,可以用来计算很多东西。比如质心,measure center $sel weight mass;RMSD:measure rmsd $sel1 $sel2;利用它还可以进行2个分子align的操作:$sel1 move [measure fit $sel1 $sel2]。
6. veclength [vecsub $com1 $com2]
VMD计算2个点之间的距离需用先把它们代表的向量相减,然后计算所产生向量的长度。
7. simdata($i.r)
这是VMD中数组的表示形式, $i来表示元素序数。
8. $var writepdb pdbfile
选择并对一个所导入的小分子进行操作之后将其写入一个PDB文件

















 3589
3589

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









