








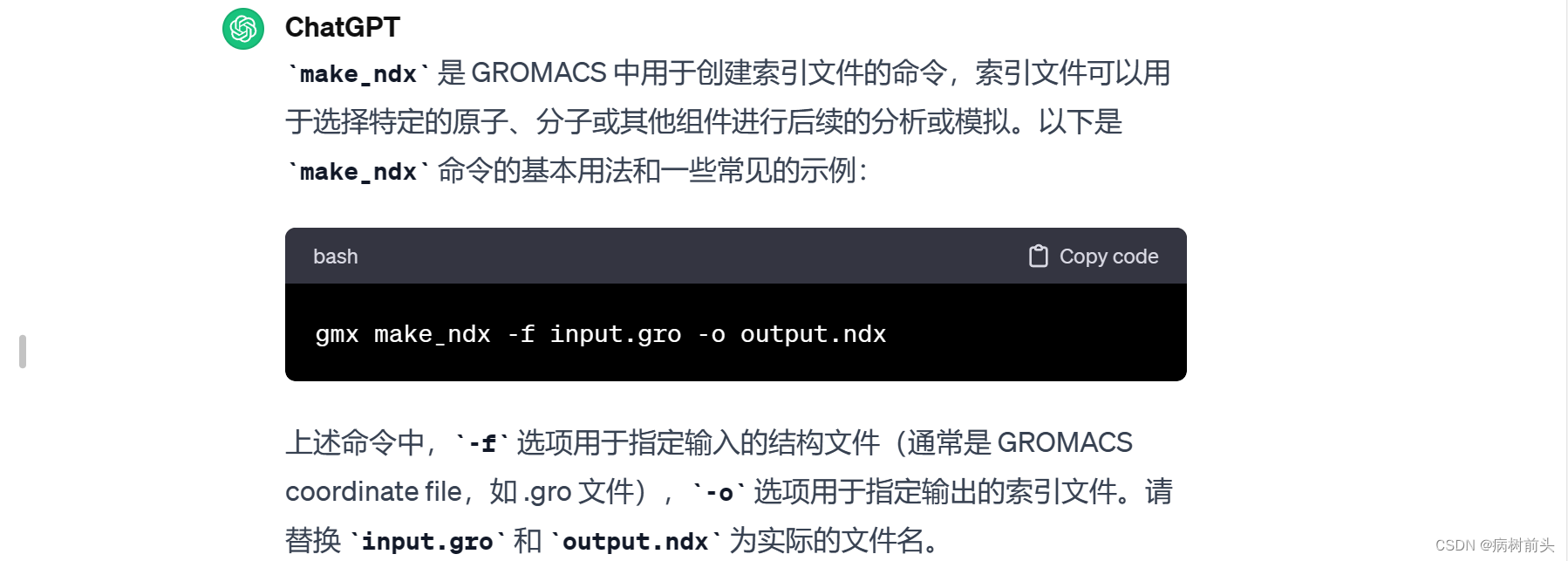
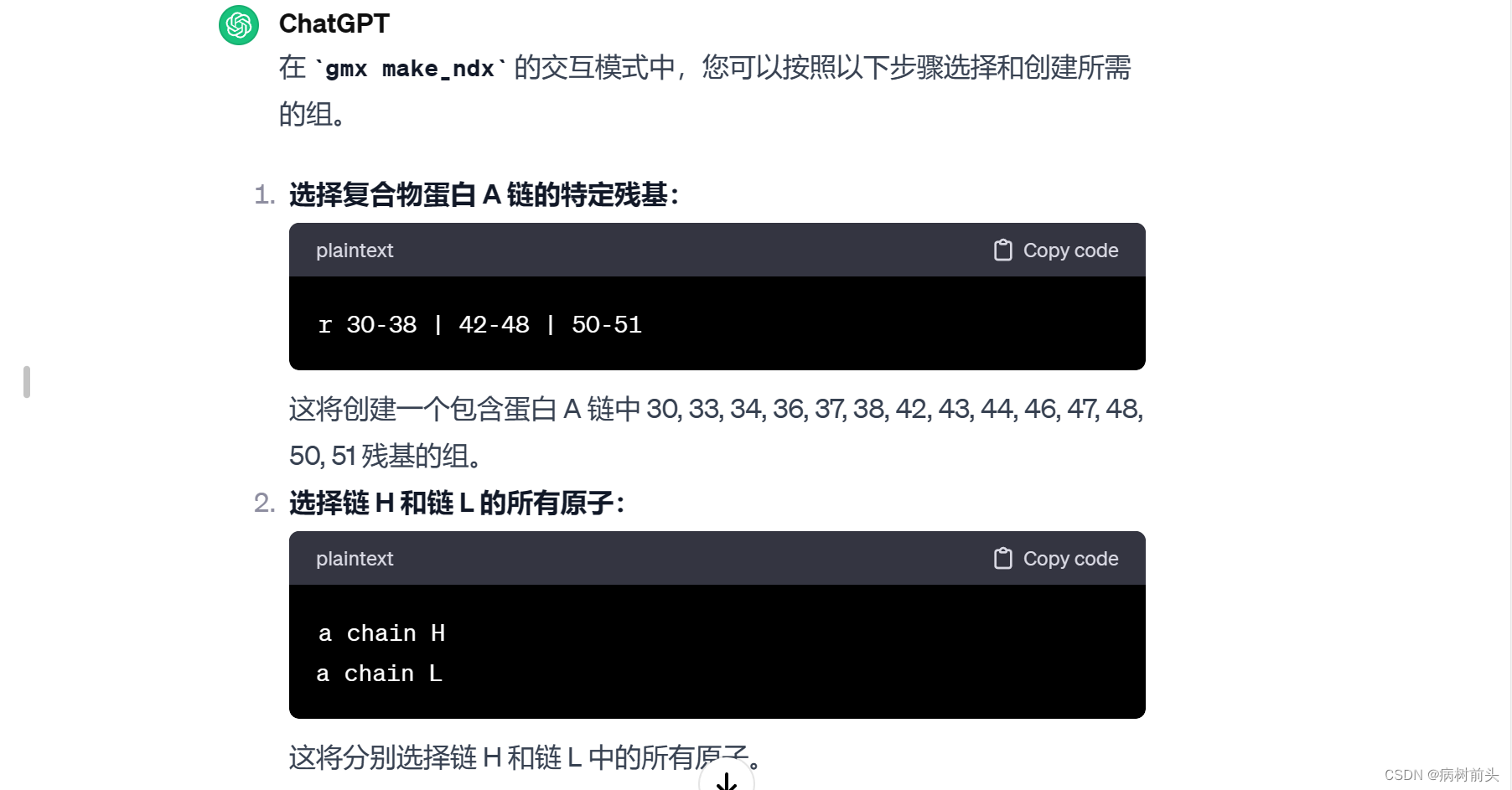
 1. 选择特定分子或原子:
1. 选择特定分子或原子:
gmx make_ndx -f input.gro -o output.ndx
这将打开交互式界面,您可以在其中选择要包含在索引文件中的分子和原子。按照提示进行操作,选择适当的分组。
2. 手动创建索引文件:
您还可以手动创建一个文本文件,其中包含要包括在索引文件中的分子和原子的详细信息。然后,您可以使用以下命令:
gmx make_ndx -f input.gro -n index.ndx < index.txt
其中 index.txt 是您手动创建的包含索引信息的文本文件。
3. 选择特定类型的原子:
gmx make_ndx -f input.gro -o output.ndx -select 'name CA'
上述命令将选择所有类型为 CA(α-碳)的原子。
4. 选择特定残基范围:
gmx make_ndx -f input.gro -o output.ndx -select 'resid 1-10'
这将选择残基编号在 1 到 10 范围内的所有原子。
5. 选择特定链:
gmx make_ndx -f input.gro -o output.ndx -select 'chain A'
上述命令将选择链标识为 A 的所有原子。
以上示例提供了一些常见的用法,您可以根据实际需要进行调整。在使用 make_ndx 命令时,请注意理解您系统的结构和拓扑信息,以便正确选择所需的分子、原子或组件。


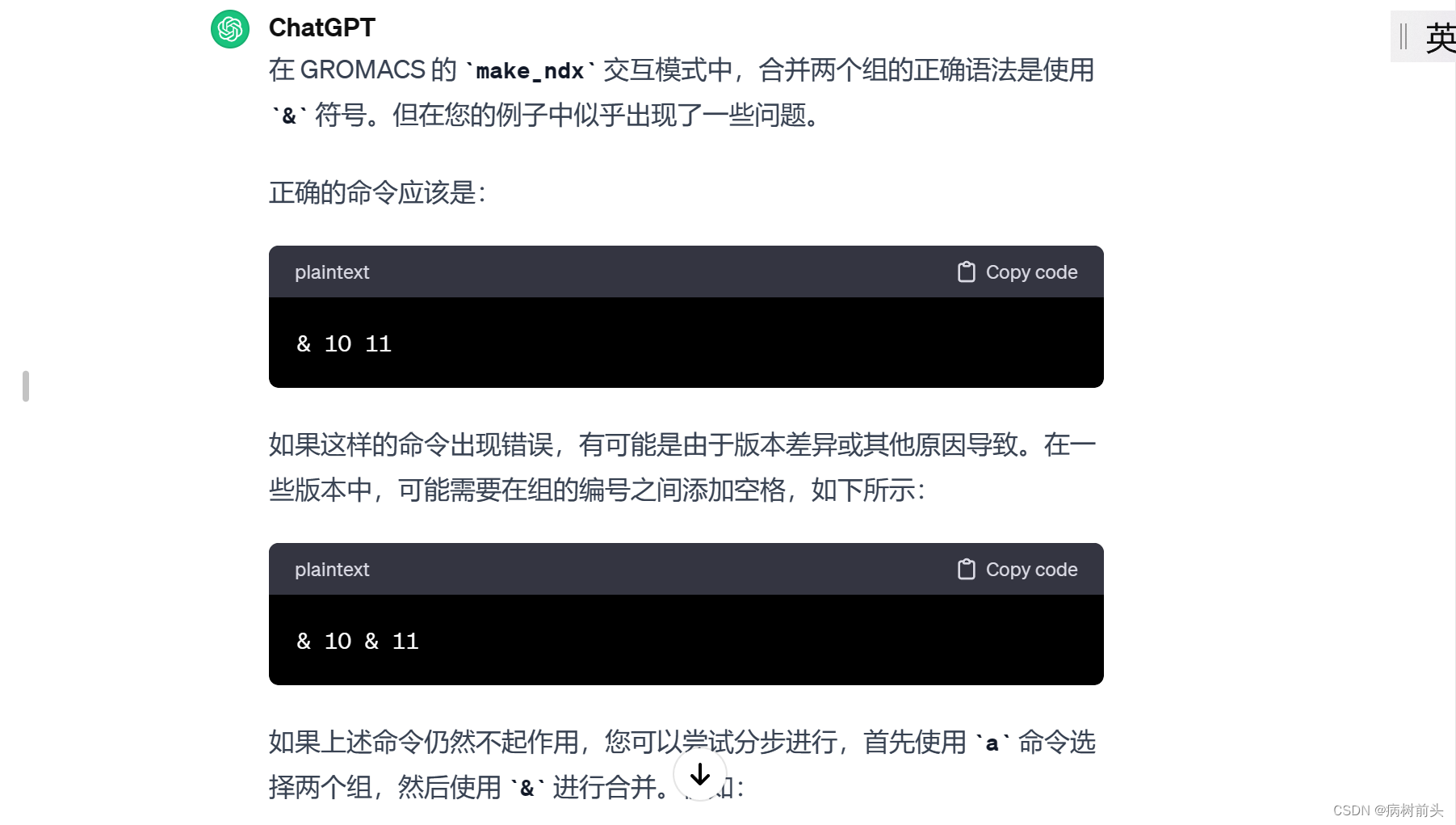
gmx make_ndx -f input.gro -o output.ndx -select 'chain A | chain B'
上述命令将选择链标识为 A 或 B 的所有原子。您可以根据实际情况修改选择条件。如果您有更复杂的选择需求,可以在 -select 语句中使用括号和逻辑运算符来组合条件。
请确保在选择条件中使用正确的拓扑信息,以便确保选择的链标识是正确的。此外,如果需要,您还可以在交互模式中手动选择链和其他组件。

 在这个例子中,index.ndx 文件定义了两个组(chain_A 和 chain_B),每个组都有一个选择条件,分别对应链 A 和链 B。在交互模式中,您可以使用这些组来进一步选择所需的分子或原子。
在这个例子中,index.ndx 文件定义了两个组(chain_A 和 chain_B),每个组都有一个选择条件,分别对应链 A 和链 B。在交互模式中,您可以使用这些组来进一步选择所需的分子或原子。
请注意,确保您的 GROMACS 版本支持 make_ndx 的 -n 选项。不同的 GROMACS 版本可能具有不同的命令行选项。
这里,input.pdb 是您的复合物蛋白结构文件,index.ndx 包含了您的选择条件。在生成的索引文件 (output.ndx) 中,您可以查看 Protein 组的编号,并使用这个编号在模拟或分析中引用该组。
如果您希望在模拟中固定这个组,可以在 mdp 文件中使用 constraints 部分,将 Protein 组中的原子位置固定。例如:

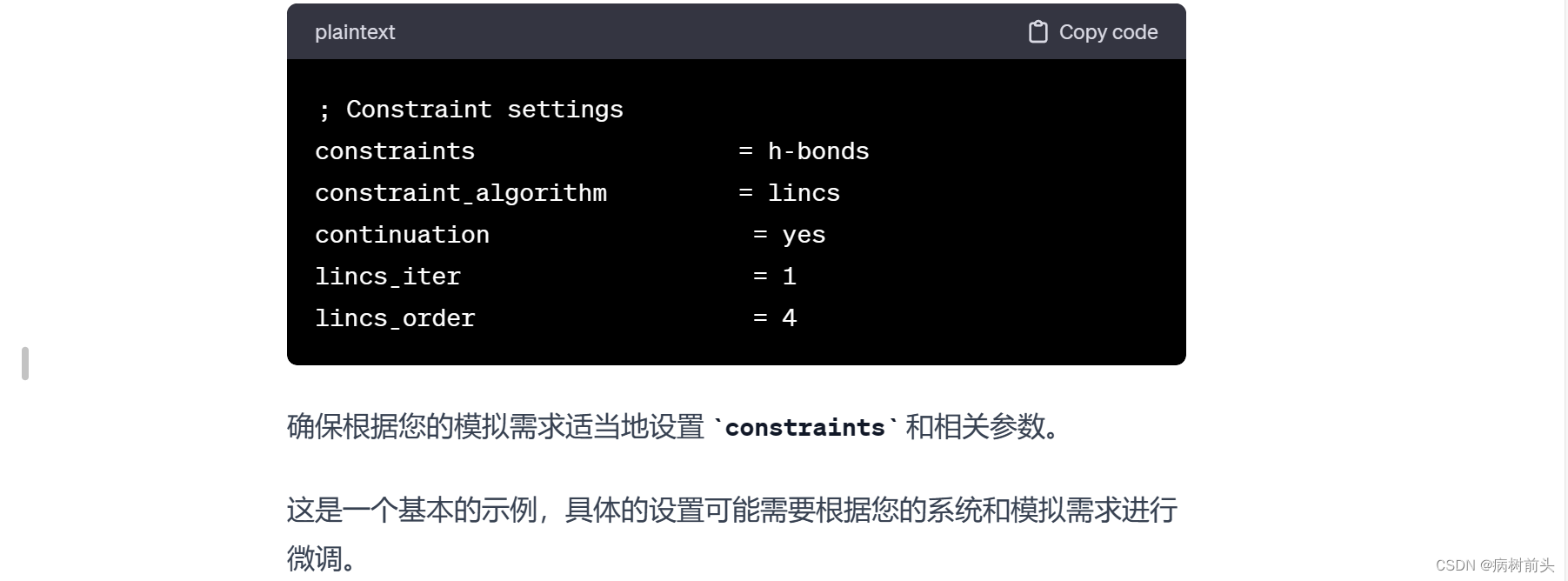










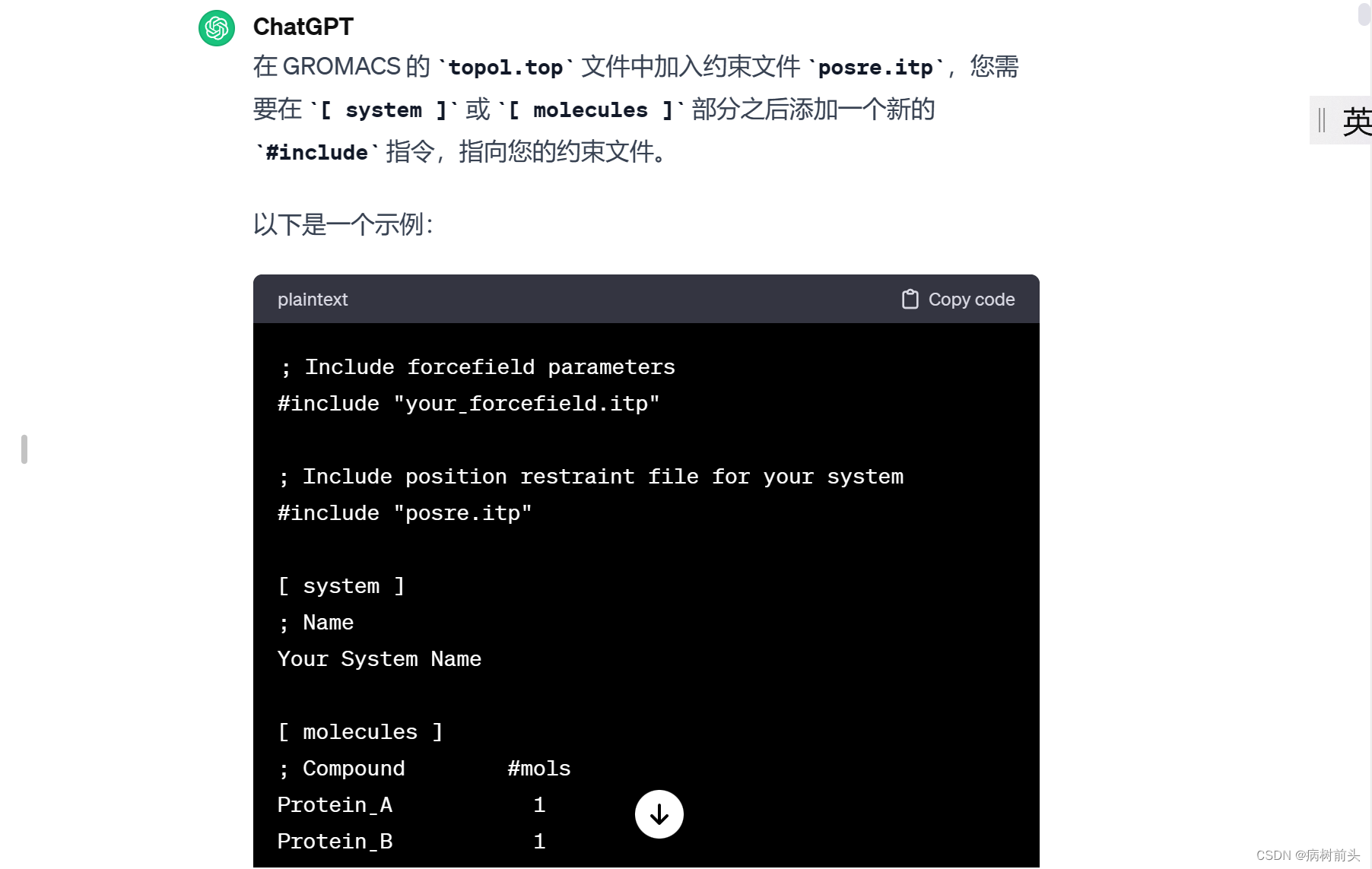 确保替换 “your_forcefield.itp” 和 “posre.itp” 为您实际使用的力场文件和位置约束文件的名称。此外,Your System Name 也应替换为您系统的名称。
确保替换 “your_forcefield.itp” 和 “posre.itp” 为您实际使用的力场文件和位置约束文件的名称。此外,Your System Name 也应替换为您系统的名称。
这里假设 posre.itp 是一个包含位置约束信息的文件,其中定义了需要进行位置约束的原子和相应的力常数。确保 posre.itp 文件位于同一目录下,或者您提供正确的路径。
在这个示例中,#include “posre.itp” 语句将 posre.itp 中的内容插入到 topol.top 文件的相应位置。这是一种组织力场和拓扑文件的常见方式,以使文件结构更清晰。


; Position restraint
define = -DPOSRES
posre_fc = 1000 ; Force constant in KJ/mol/nm^2
这些设置的含义如下:
define = -DPOSRES: 这个选项告诉 GROMACS 启用位置约束。定义 POSRES 会在仿真中引入位置约束。
posre_fc = 1000: 这是位置约束的力常数,表示施加在原子上的力的大小。在这个例子中,力常数是 1000 KJ/mol/nm^2。您可以根据您的系统性质和仿真需求来调整这个值。通常,较小的值会引入较弱的位置约束,较大的值会引入较强的位置约束。
确保将这些设置添加到 md.mdp 文件的适当位置,通常是在文件的 [constraints] 或 [integrator] 部分之后。添加这些设置后,您可以使用这个 md.mdp 文件运行 GROMACS 模拟,系统将受到定义的位置约束。

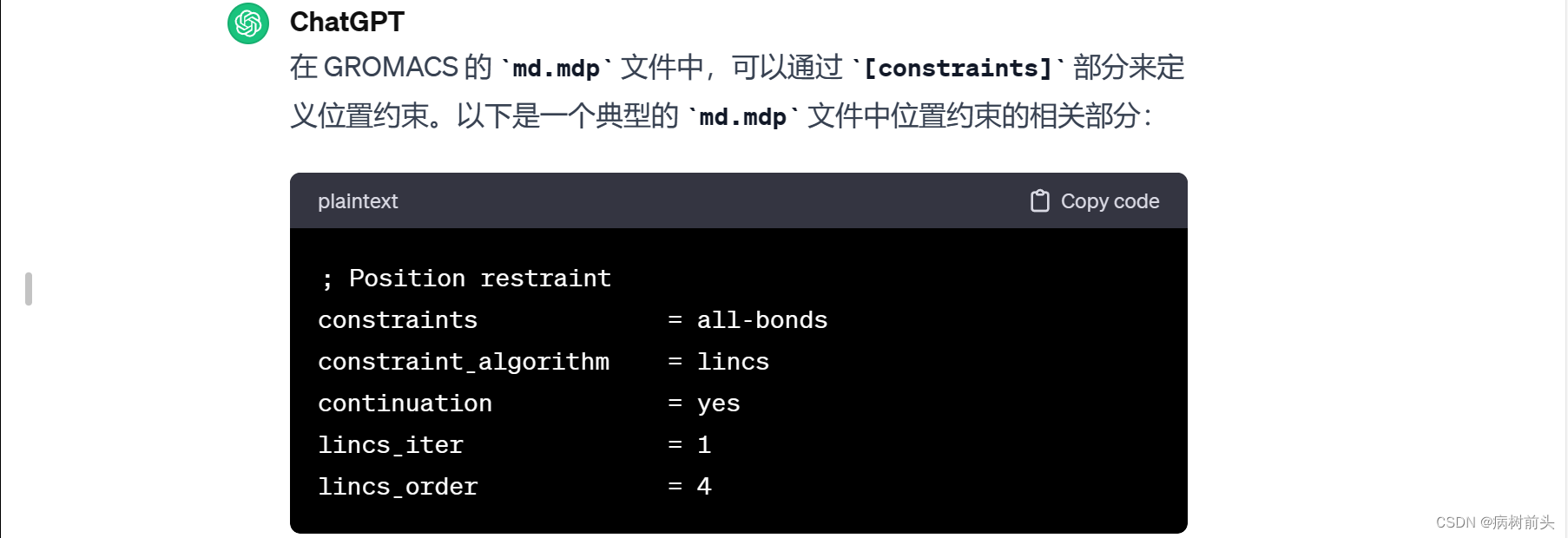 这里,constraints = all-bonds 意味着使用所有键的约束,但您也可以选择其他的约束方式,具体取决于您的系统和仿真需求。一些可能的选项包括:
这里,constraints = all-bonds 意味着使用所有键的约束,但您也可以选择其他的约束方式,具体取决于您的系统和仿真需求。一些可能的选项包括:
none: 不使用任何约束。
h-bonds: 仅使用氢键约束。
all-bonds: 使用所有键的约束。
而 constraint_algorithm = lincs 表示采用线性约束算法(LINCS)。这是一种常见的约束算法,适用于大多数分子模拟系统。lincs_iter 和 lincs_order 是相关参数,您可以根据系统需要进行调整。
请根据您的系统性质和仿真需求,选择适当的约束方式和参数。如果您使用了 define = -DPOSRES,在这种情况下,位置约束将自动启用,并且不需要额外的 constraints 部分。确保将这些设置添加到 md.mdp 文件的适当位置,通常是在文件的 [constraints] 或 [integrator] 部分之后。


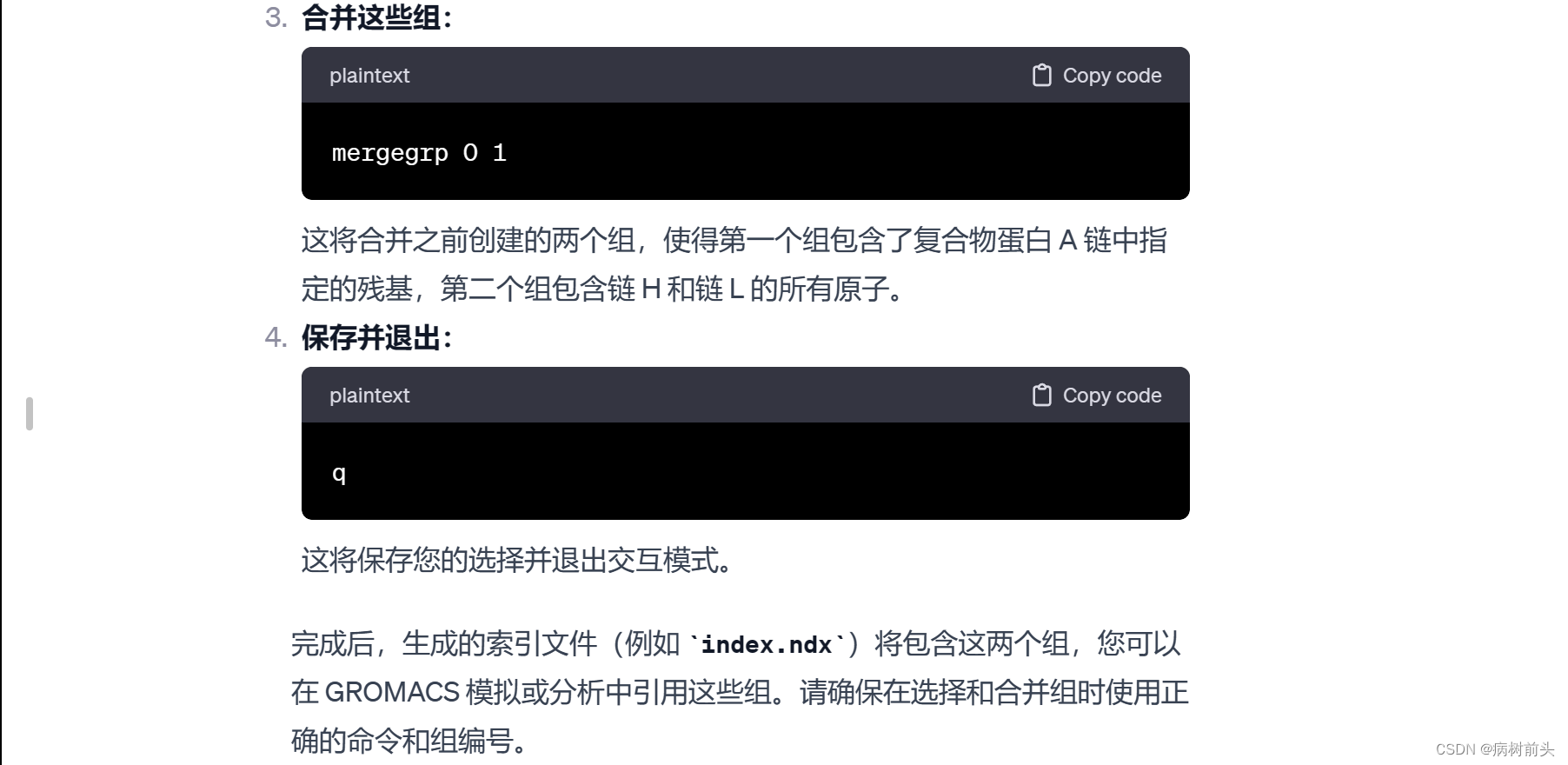

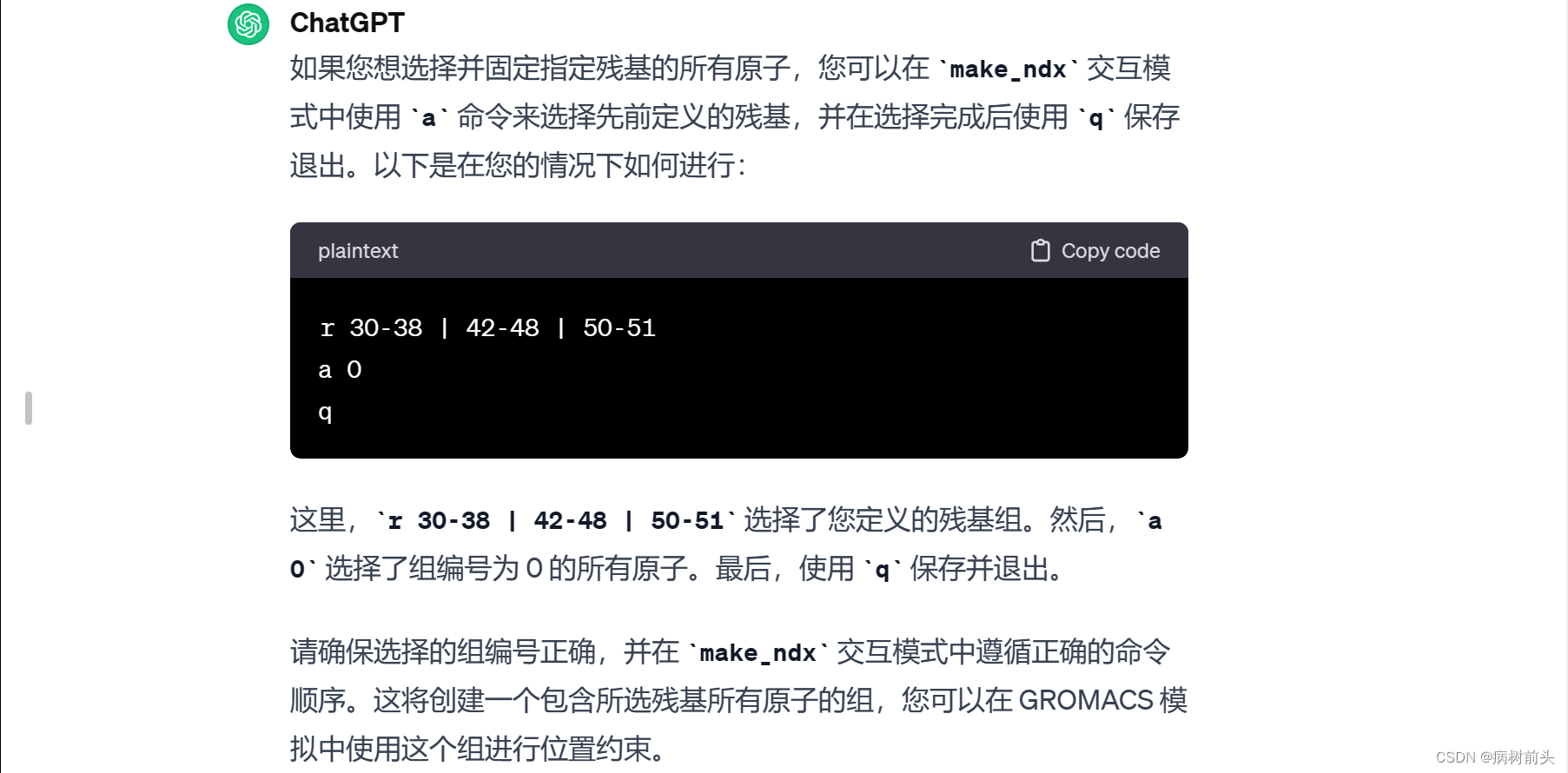

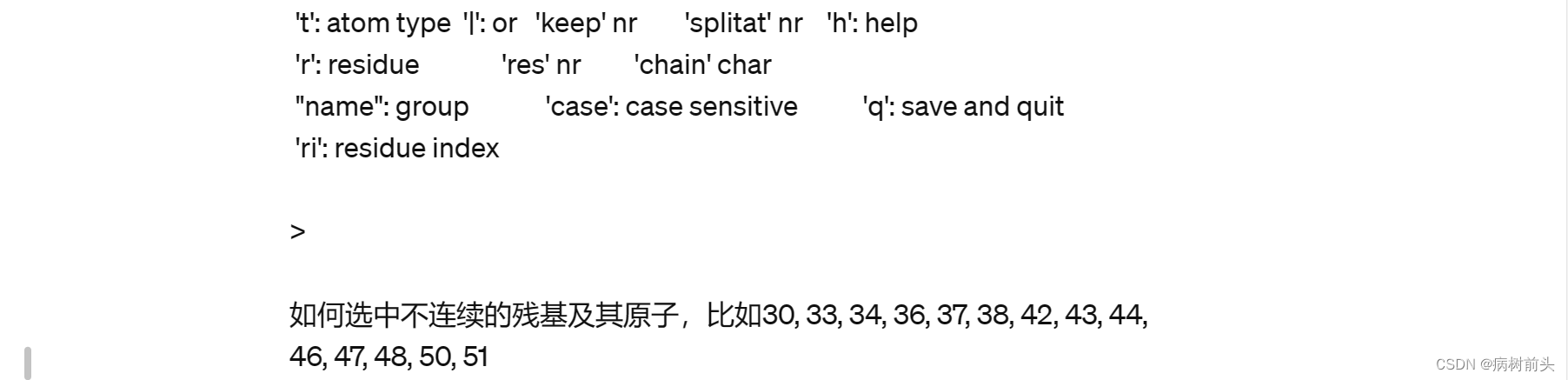
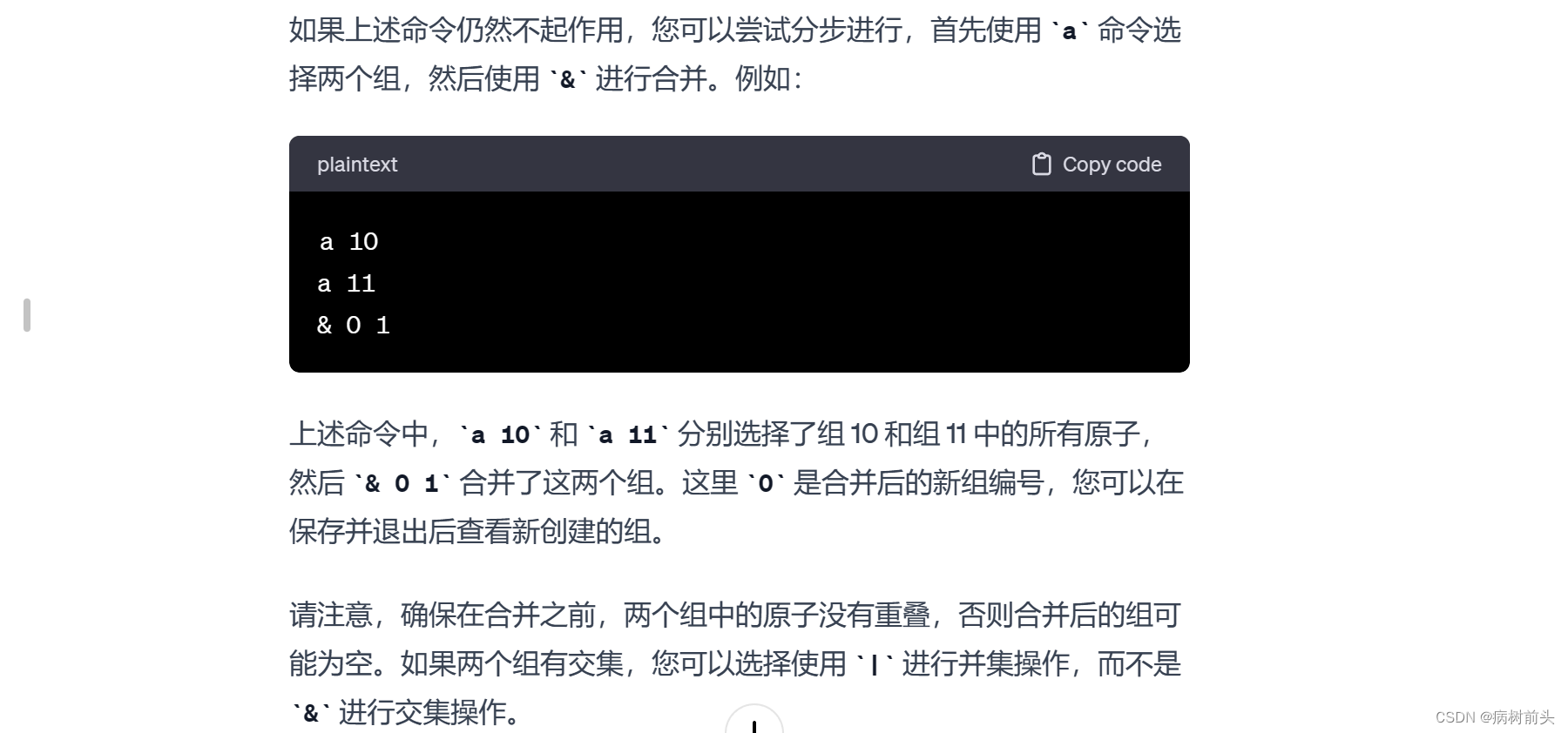
 这里,a 0 表示选择组编号为 0 的所有原子。最后,使用 q 保存并退出。
这里,a 0 表示选择组编号为 0 的所有原子。最后,使用 q 保存并退出。
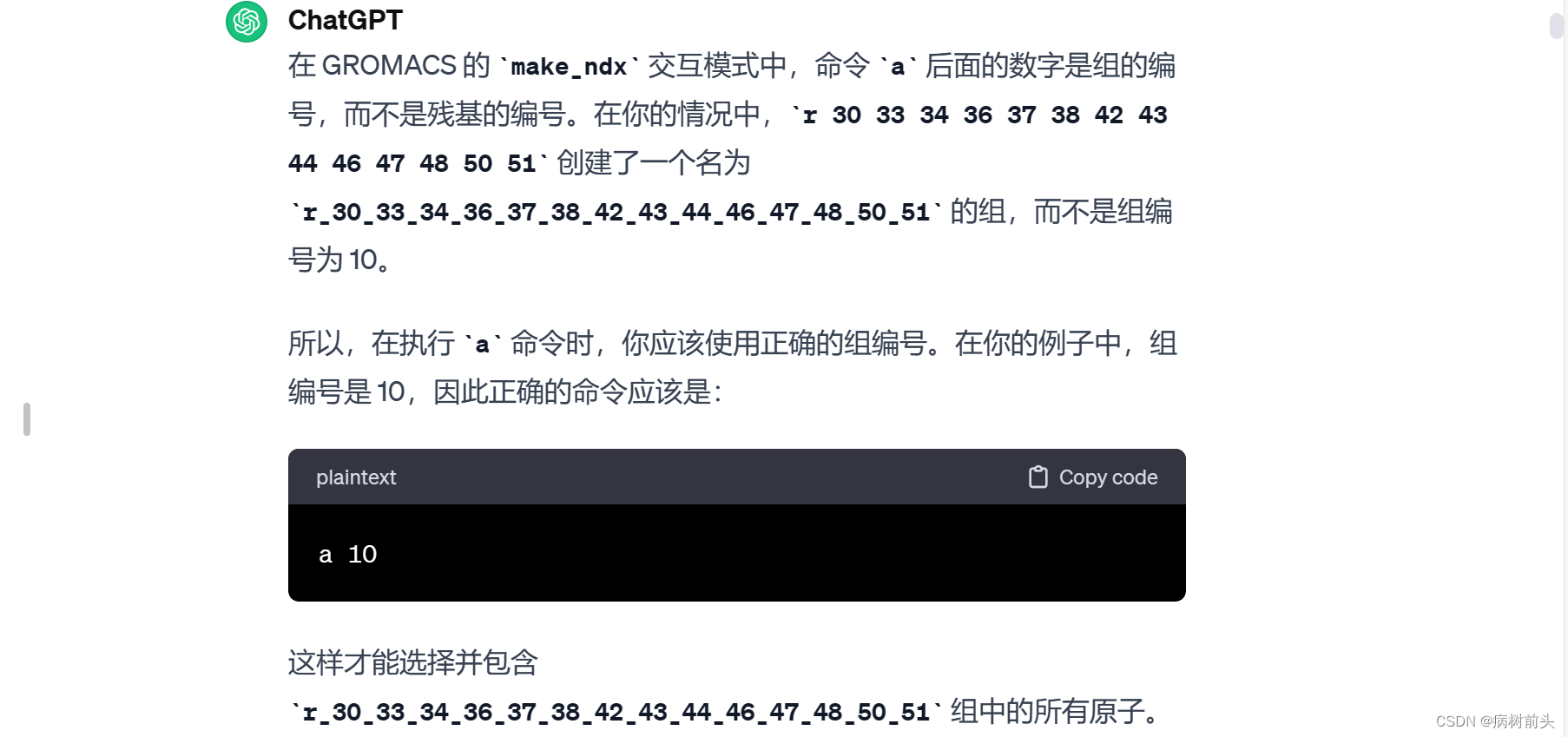
确保在选择残基时使用正确的命令和语法,并根据需要使用其他命令选择或操作不同的组。


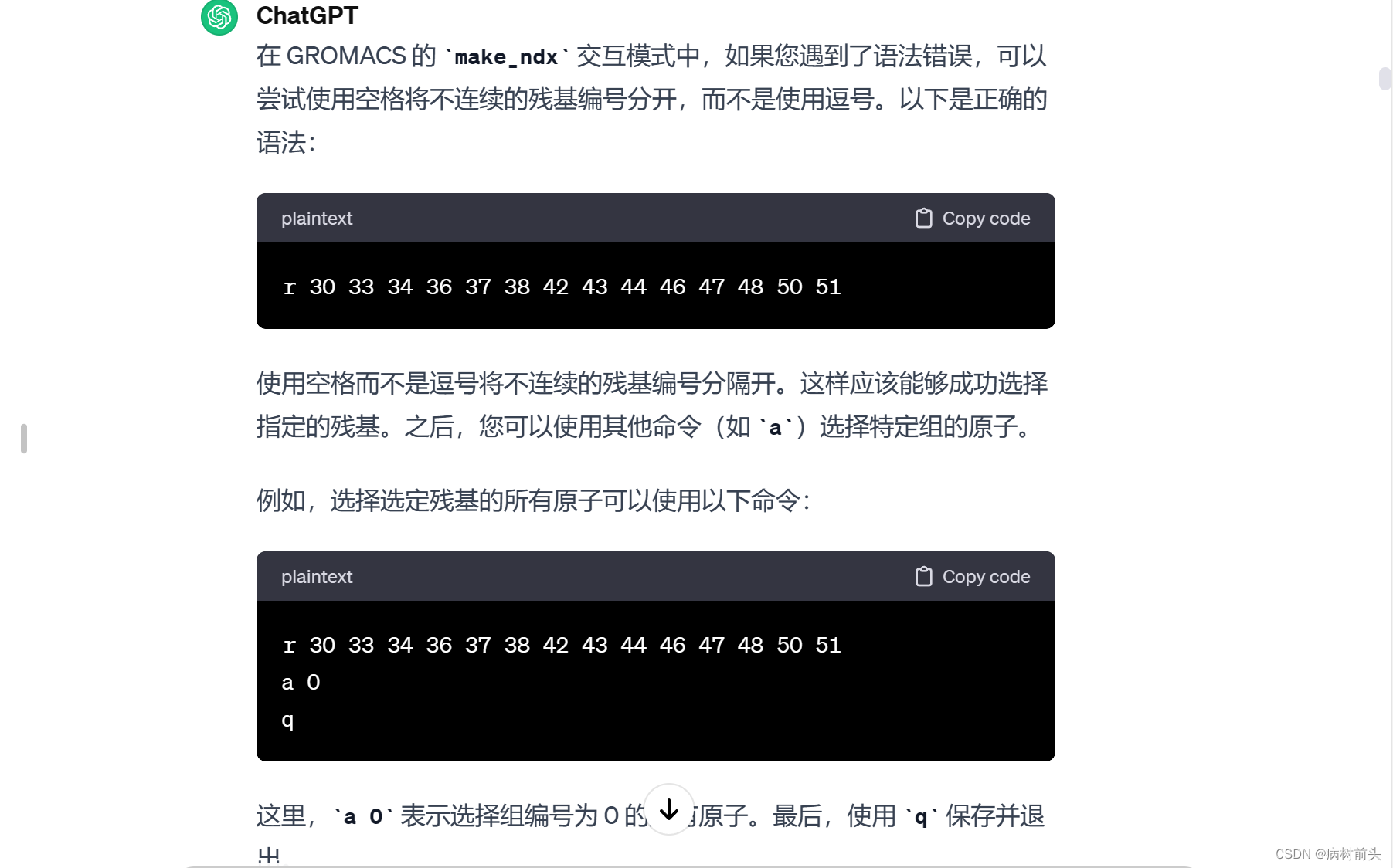 请确保在选择残基时使用正确的命令和语法,并根据需要使用其他命令选择或操作不同的组。
请确保在选择残基时使用正确的命令和语法,并根据需要使用其他命令选择或操作不同的组。


















 673
673

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









