







欢迎关注bioinfor 生信云!有一起想做公众号的朋友欢迎联系我!
上一篇推文讲了bulk RNA-Seq 的比对到参考基因组部分,接下来就是基因表达定量了。计算表达定量可以用StringTie、Htseq-cout、featureCounts,这里推荐使用featureCounts。featureCounts 是Rsubread 软件包里的一个命令,所以安装R版本Rsubread的即可。这里使用了一个脚本,在运行featureCounts 的同时,计算FPKM、TPM。
安装相关的R包
if(!requireNamespace("BiocManager", quietly = TURE))
install.packages("BiocManager")
BiocManager::install("Rsubread")
BiocManager::install("limma")
BiocManager::install("edgeR")
featureCounts.R
# Create a parser
p <- arg_parser("run featureCounts and calculate FPKM/TPM")
# Add command line arguments
p <- add_argument(p, "--bam", help="input: bam file", type="character")
p <- add_argument(p, "--gtf", help="input: gtf file", type="character")
p <- add_argument(p, "--output", help="output prefix", type="character")
# Parse the command line arguments
argv <- parse_args(p)
library(Rsubread)
library(limma)
library(edgeR)
bamFile <- argv$bam
gtfFile <- argv$gtf
nthreads <- 1
outFilePref <- argv$output
outStatsFilePath <- paste(outFilePref, '.log', sep = '');
outCountsFilePath <- paste(outFilePref, '.count', sep = '');
fCountsList = featureCounts(bamFile, annot.ext=gtfFile, isGTFAnnotationFile=TRUE, nthreads=nthreads, isPairedEnd=TRUE)
dgeList = DGEList(counts=fCountsList$counts, genes=fCountsList$annotation)
fpkm = rpkm(dgeList, dgeList$genes$Length)
tpm = exp(log(fpkm) - log(sum(fpkm)) + log(1e6))
write.table(fCountsList$stat, outStatsFilePath, sep="\t", col.names=FALSE, row.names=FALSE, quote=FALSE)
脚本用法
输入:bam文件,gtf文件
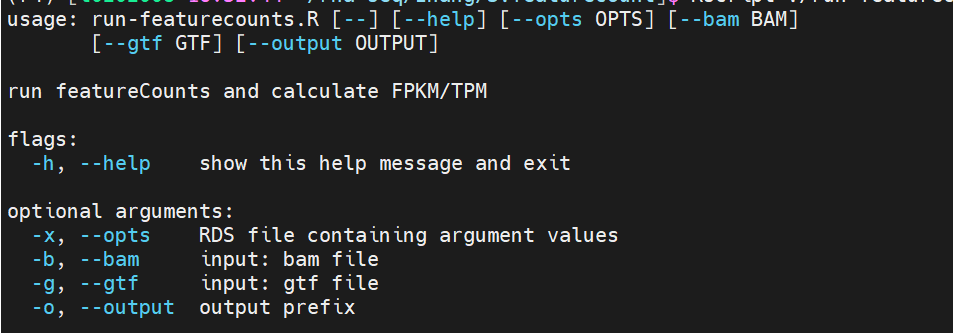
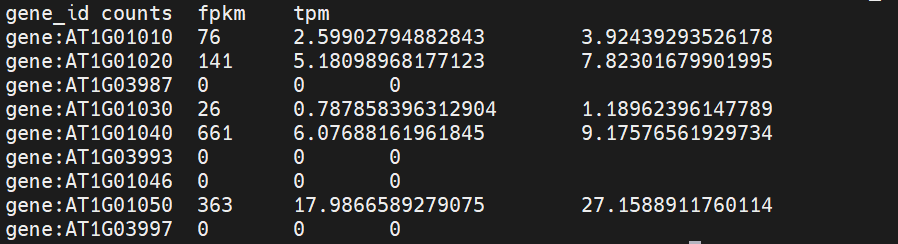
结果输出
.count结尾的表达定量文件
.log结尾的日志文件
Assigned 17323283 #基因结构上的reads
Unassigned_Unmapped 2909094 #未比对上的reads
Unassigned_Read_Type 0
Unassigned_Singleton 0
Unassigned_MappingQuality 0
Unassigned_Chimera 0
Unassigned_FragmentLength 0
Unassigned_Duplicate 0
Unassigned_MultiMapping 0
Unassigned_Secondary 0
Unassigned_NonSplit 0
Unassigned_NoFeatures 408001 #未落在基因结构上
Unassigned_Overlapping_Length 0
Unassigned_Ambiguity 1180806 #无法确定分配到哪个基因
















 3万+
3万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











