







欢迎关注bioinfor 生信云!有一起想做公众号的朋友欢迎联系我!
我们做完了上游的基础分析之后,接下来就是数据挖掘了。我们先准备数据挖掘的三张表。
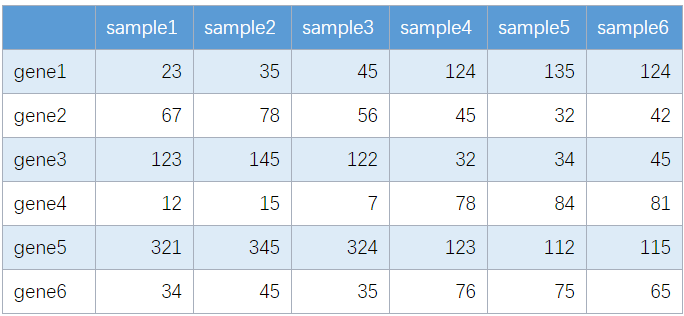
表达矩阵(gene_exp)
每一行是一个基因,每一列是一个样本,需要对数据进行标准化。
标准化之前的read count 矩阵,用于差异表达分析
标准化之后的TPM/FPKM 矩阵,用于其他分析(PCA分析、聚类分析等等)
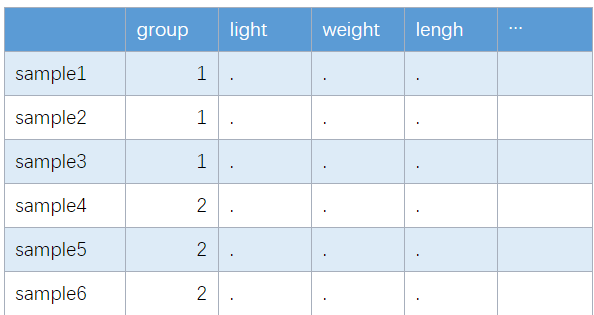
样本信息表(sample_info)
每一行是一个样本,每一列是一个表型特征(光照、地上生物量、茎长等等),可以和基因进行关联分析
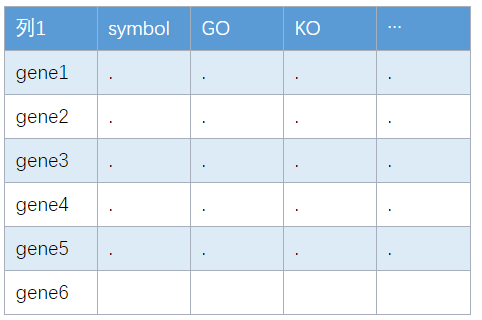
基因信息表(gene_info)
每一行是一个基因,每一列是该基因的信息(symbol、KO、GO),基因的信息可以通过eggnog-mapper在线网站注释得到。
准备好这三张表之后,我们就可以去画图啦,下期见。

















 3万+
3万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











