







在高级篇1中回顾了用于解析细胞间通讯网络的工具——CellChat。
接下来将回顾学习CytoTRACE2和monocle3两个工具,CytoTRACE2通过预测细胞发育潜能分数和分类,辅助研究者定位发育轨迹的起始点;而monocle3则基于图论算法构建细胞伪时间分化轨迹,动态模拟细胞状态沿时间轴的演化路径,揭示分化分支、过渡态细胞及关键驱动基因。
本次内容涉及到的工程文件可通过网盘获得:中级篇2,链接: https://pan.baidu.com/s/1y-HHLXoXsJbgWKCdz26-gQ 提取码: yx93 。
此外,可以向“生信技能树”公众号发送关键词‘单细胞’,直接获取Seurat V5版本的完整代码。
CytoTRACE2
CytoTRACE2是一种基于单细胞RNA测序数据预测细胞潜能类别及绝对发育潜力的计算方法。其定义的潜能类别将细胞分为多个层次:1. 根据细胞的分化潜能对细胞进行分类:totipotent(多谱系分化的全能)—pluripotent(多谱系分化的多能)—multipotent(谱系限制分化的多能)—oligopoten(谱系限制分化的寡能)—unipotent(谱系限制分化的单能)—differentiated(分化); 2. 该方法通过预测的潜能分数(范围0到1)提供连续发育潜力评估,其中0(differentiated)代表分化,1(totipotent)代表全能。3.其核心是一个新型可解释的深度学习框架,该框架基于覆盖发育全谱系的34个人类和小鼠单细胞数据集(涵盖24种组织类型)训练验证,可学习每个潜能类别的基因表达程序,并通过校准不同细胞发育起源的输出结果,实现跨数据集比较发育潜力。
Monocle3
Monocle3拟时序分析采用UMAP进行非线性降维,将高维基因表达数据映射到低维空间以保留细胞间的相似性,随后结合Louvain社区检测算法识别细胞群体,并将相邻群体连接形成“超级组”,最终通过图论方法推断出细胞状态的拓扑结构,如线性、分支或循环轨迹。相较于Monocle2(基于反向图嵌入和生成树模型),Monocle3提升了对复杂轨迹的处理能力,优化了计算效率以支持大规模数据集分析。研究者可通过伪时间排序量化细胞在轨迹中的位置,识别关键驱动基因,并可视化分化路径。
同时还需要学习一下拟时序(pseudotime)这个概念,其是一种衡量单个细胞在某一过程(如细胞分化)中进展程度的方式。在许多生物过程中,细胞并不会完全同步地进行。在对细胞分化等过程进行的单细胞表达研究中,即使在完全相同的时间点捕获的细胞,在分化程度上也是十分多样的。也就是说,在同一时间捕获的一群细胞中,有些细胞可能已经分化到了很后阶段,而另一些细胞可能尚未开始这一过程。这种不同步现象在试图理解细胞从一种状态过渡到另一种状态时所发生的调控变化顺序时,会造成很大困难。若以时间为基准追踪基因表达的变化,会使基因动力学显得非常“压缩”(个人理解是如果每个细胞都按假定的时间去设定的话数据会变得复杂和可读性不佳),且基因的表达表现出很高的表观变异性。Monocle通过根据学习到的轨迹对每个细胞的分化进展进行排序,从而缓解了由于不同步所带来的问题(个人理解是按照基因的表达情况对细胞进行排序)。Monocle不是将表达变化视为时间的函数,而是作为沿轨迹进展的函数来跟踪变化称其为“拟时”(一旦对细胞进行排序之后,就可以在图上找到细胞与细胞之间的距离,用距离差异进行"拟时")。拟时是一种抽象的进展单位:它只是细胞与轨迹起始点之间的距离,该距离沿轨迹的最短路径计算。轨迹的总长度是通过细胞从起始状态到最终状态在转录上的总变化量来定义的。
Cytotrace2分析流程
1.导入
rm(list=ls())
source('scRNA_scripts/lib.R')
source('scRNA_scripts/mycolors.R')
library(tidyverse)
library(CytoTRACE2)
library(Seurat)
library(paletteer)
library(BiocParallel)
register(MulticoreParam(workers = 8, progressbar = TRUE))
sub_data <- qread("./9-CD4+T/CD4+t_final.qs")
# check一下
DimPlot(sub_data,pt.size = 0.8,group.by = "celltype",label = T)
dir.create("./13-monocle")
setwd("./13-monocle")
2.数据预处理
cytotrace2_res <- cytotrace2(sub_data, #seurat对象
is_seurat = TRUE,
slot_type = "counts", #counts和data都可以
species = 'human')#物种要选择,默认是小鼠
class(cytotrace2_res)
3.可视化
annotation <- data.frame(phenotype = sub_data@meta.data$celltype) %>%
set_rownames(., colnames(sub_data))
# plotting-一次性生成多个图,然后储存在一个list,用$查看即可
plots <- plotData(cytotrace2_result = cytotrace2_res,
annotation = annotation,
is_seurat = TRUE)
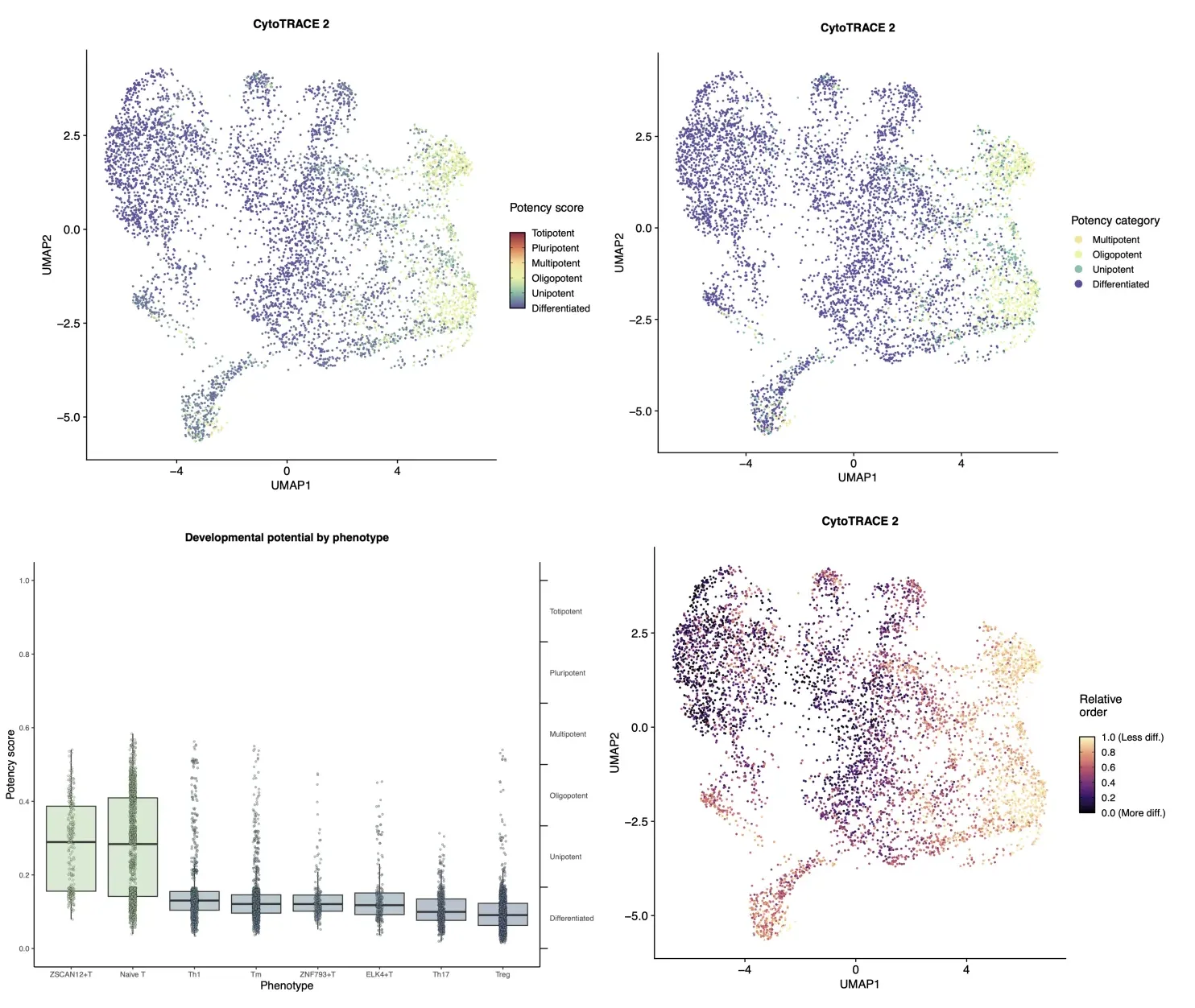
plots$CytoTRACE2_UMAP
ggsave("CytoTRACE2_UMAP.pdf", width = 9, height = 7, dpi = 300)
plots$CytoTRACE2_Potency_UMAP
ggsave("CytoTRACE2_Potency_UMAP.pdf", width = 9, height = 7, dpi = 300)
plots$CytoTRACE2_Relative_UMAP
ggsave("CytoTRACE2_Relative_UMAP.pdf", width = 9, height = 7, dpi = 300)
plots$Phenotype_UMAP
ggsave("Phenotype_UMAP.pdf", width = 9, height = 7, dpi = 300)
plots$CytoTRACE2_Boxplot_byPheno
ggsave("CytoTRACE2_Boxplot_byPheno.pdf", width = 9, height = 7, dpi = 300)
根据当前分析结果,ZSCAN12+T细胞群展现出最显著的全能性(Totipotent)倾向,而其余细胞群基本呈现分化状态(Differentiated)特征。需要说明的是,本次分析以流程演示为核心目标,侧重方法学验证而非生物学机制解读。若在实际科研场景中开展此类分析,需严格验证各细胞群的功能合理性(例如ZSCAN12是否确为全能性标志基因、分化状态是否符合组织特异性发育轨迹等),避免仅依赖算法预测而忽略生物学背景的交叉验证。
monocle3分析流程
1.导入
rm(list = ls())
source('scRNA_scripts/lib.R')
source('scRNA_scripts/mycolors.R')
library(paletteer)
library(Seurat)
library(monocle3)
library(dplyr)
library(BiocParallel)
register(MulticoreParam(workers = 4, progressbar = TRUE))
sub_data <- qread("./9-CD4+T/CD4+t_final.qs")
# check一下
DimPlot(sub_data,pt.size = 0.8,group.by = "celltype",label = T)
dir.create("./13-monocle")
setwd("./13-monocle")
2.数据预处理
scRNA <- sub_data
Idents(scRNA) <- scRNA$celltype
levels(Idents(scRNA)) #打出来细胞类型供复制
# [1] "Th1" "Treg" "Tm" "Naive T" "Th17" "ZNF793+T" "ELK4+T"
# [8] "ZSCAN12+T"
# 根据CytoTRACE2/先验知识进行重设等级
scRNA$celltype <- factor(scRNA$celltype,levels = c("ZSCAN12+T","Naive T","Th1","Tm",
"ZNF793+T","ELK4+T","Th17","Treg"))
Idents(scRNA) <- scRNA$celltype
## 提取数据
expression_matrix <- GetAssayData(scRNA, assay = 'RNA',slot = 'counts')
cell_metadata <- scRNA@meta.data
gene_annotation <- data.frame(gene_short_name = rownames(expression_matrix))
rownames(gene_annotation) <- rownames(expression_matrix)
3.构建细胞轨迹
cds <- new_cell_data_set(expression_matrix,
cell_metadata = cell_metadata,
gene_metadata = gene_annotation)
4.monocle3分析前预处理
# 归一化/预处理数据
# 这一步使用的PCA分析,dim数代表纳入的PCA数量
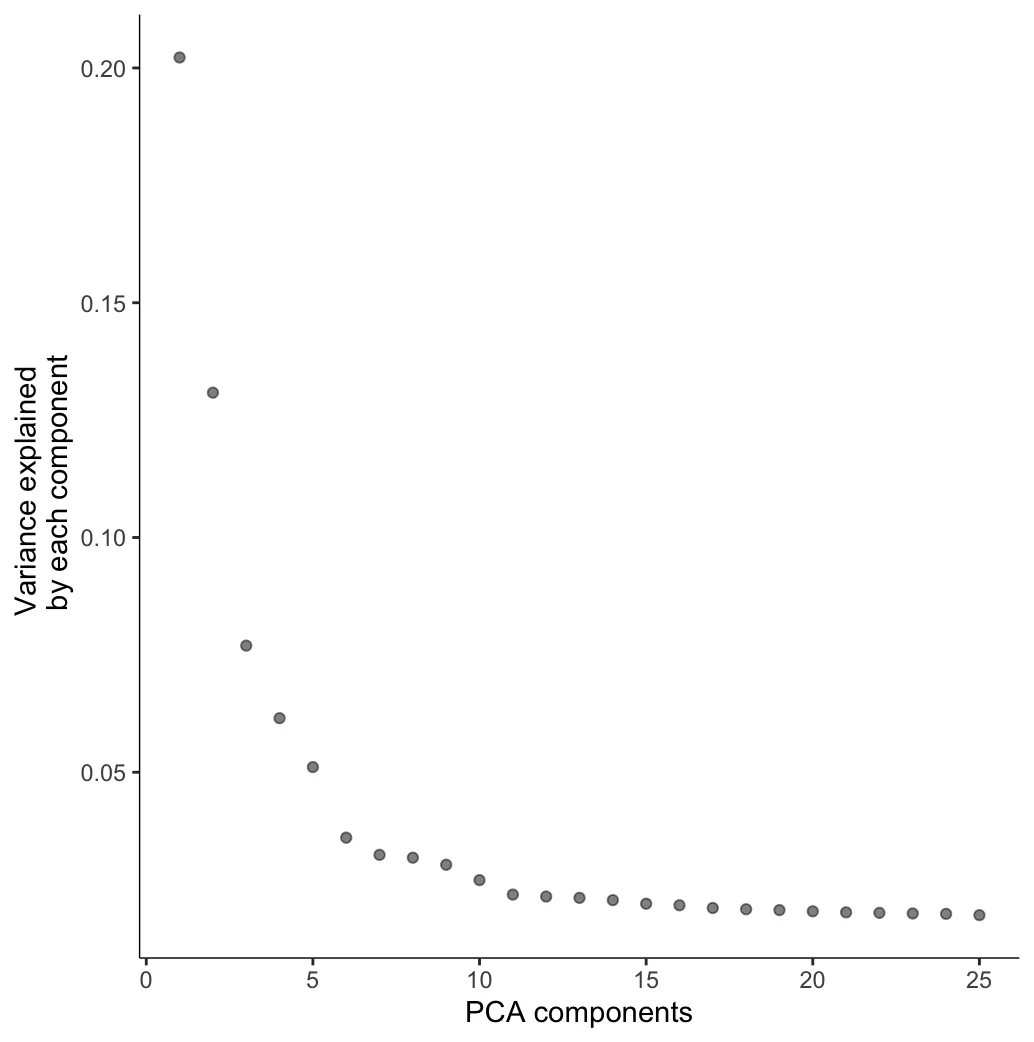
cds <- preprocess_cds(cds, num_dim = 25)
# 这个函数用于确认设定的dim数是否足够代表主要变异
plot_pc_variance_explained(cds)
# 可选(去批次处理)
#cds <- align_cds(cds, num_dim = 100, alignment_group = "GSE_num")
选择一个能够保证大部分Variance都纳入的PCA componets值。
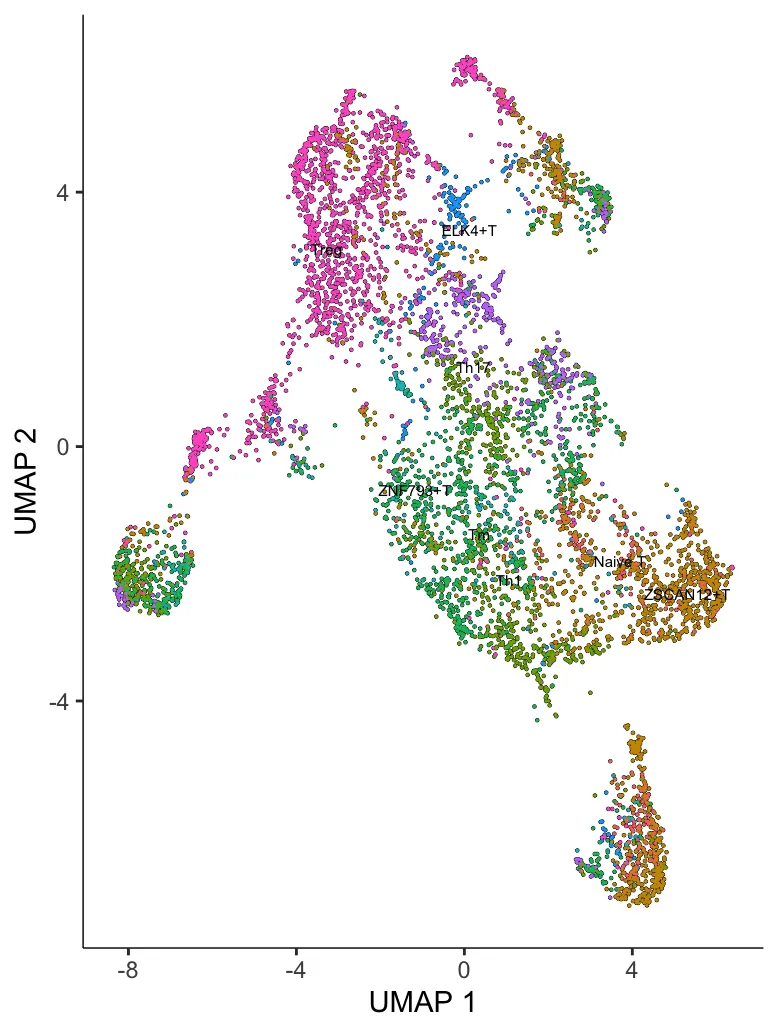
5.降维并可视化
# 降维聚类,可选择UMAP、PCA或者TSNE
cds <- reduce_dimension(cds,reduction_method='UMAP',preprocess_method = 'PCA')
plot_cells(cds, label_groups_by_cluster=FALSE, color_cells_by = "celltype")
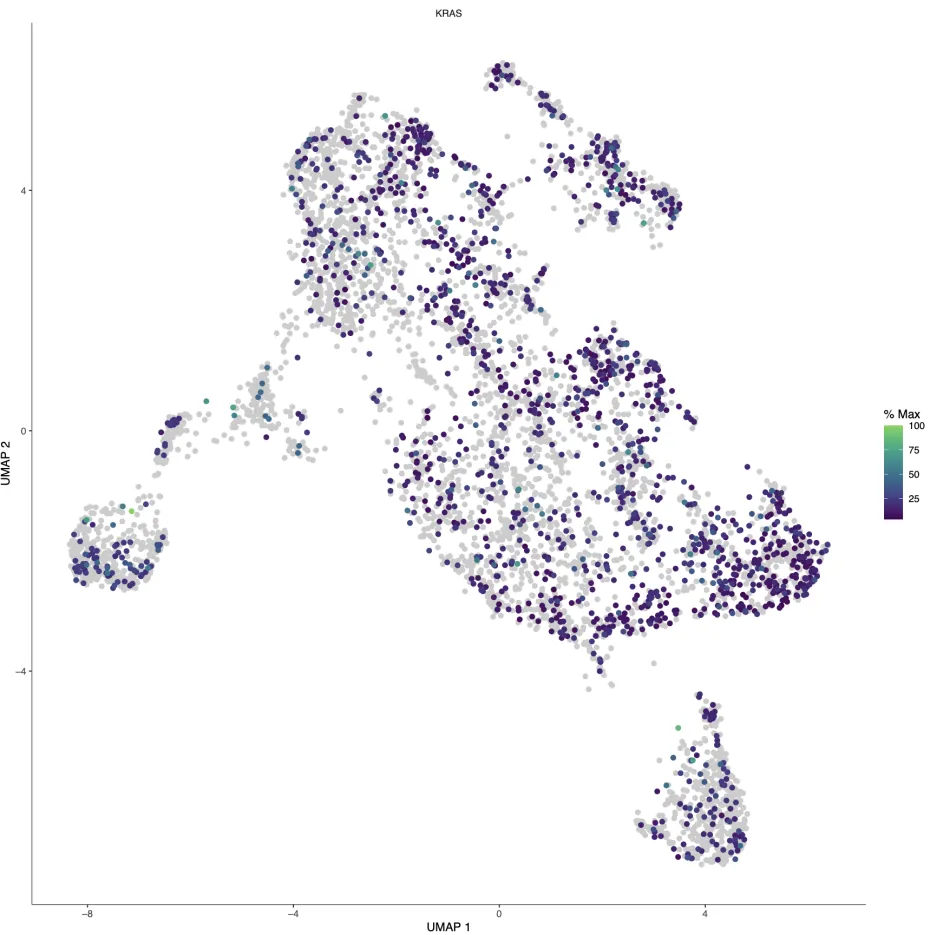
基因/轨迹共定位
这里可以看基因和轨迹的共定位情况
modelgenes <- "KRAS"
plot_cells(cds,
cell_size=1.5,group_label_size=4,
genes=modelgenes,
label_cell_groups=FALSE,
show_trajectory_graph=FALSE)
ggsave("genes_show.pdf",width = 12,height = 12)
6.细胞聚类
cds <- cluster_cells(cds) #cluster your cells
plot_cells(cds, color_cells_by = "celltype")
7.轨迹分析
# 轨迹推断
cds <- learn_graph(cds,verbose=T,
use_partition=T, #默认是T,T时是顾及全局的情况
)
plot_cells(cds,
color_cells_by = 'celltype',
label_groups_by_cluster=FALSE,
cell_size=1,group_label_size=4,
label_leaves= F, # 是否显示不同细胞结局
label_branch_points=F, # 是否显示不同的分支节点
trajectory_graph_color='#023858',
trajectory_graph_segment_size = 1)
接下来按照CytoTRACE2中的结果设定细胞发育的起点。
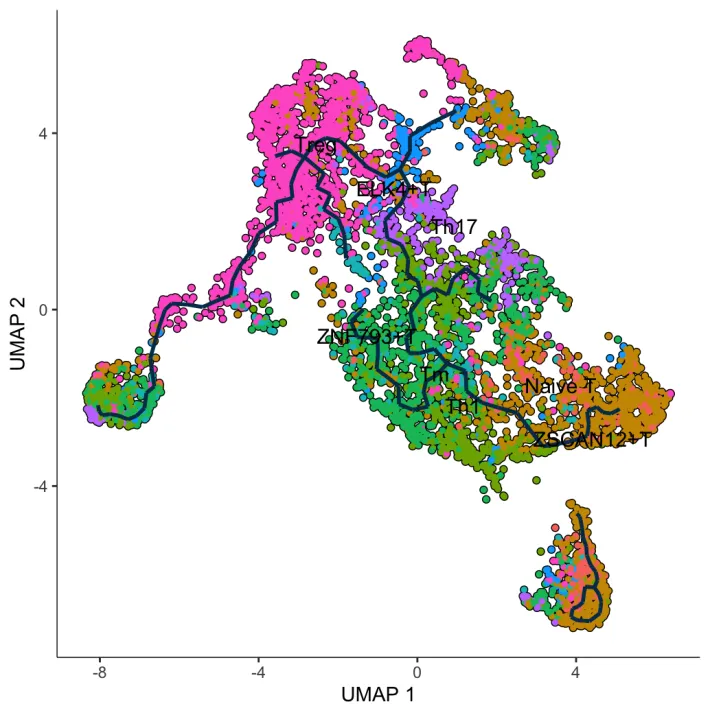
8.定义起点-轨迹可视化
#定义root cell, 推断拟时方向
# 网页自定
cds <- order_cells(cds)
# 代码定
# a helper function to identify the root principal points:
## 指定初始细胞群(ZSCAN12+T)和细胞亚群的列名(celltype)
get_earliest_principal_node <- function(cds, time_bin="ZSCAN12+T"){
cell_ids <- which(colData(cds)[, "celltype"] == time_bin)
closest_vertex <-
cds@principal_graph_aux[["UMAP"]]$pr_graph_cell_proj_closest_vertex
closest_vertex <- as.matrix(closest_vertex[colnames(cds), ])
root_pr_nodes <-
igraph::V(principal_graph(cds)[["UMAP"]])$name[as.numeric(names
(which.max(table(closest_vertex[cell_ids,]))))]
root_pr_nodes
}
cds <- order_cells(cds, root_pr_nodes=get_earliest_principal_node(cds))
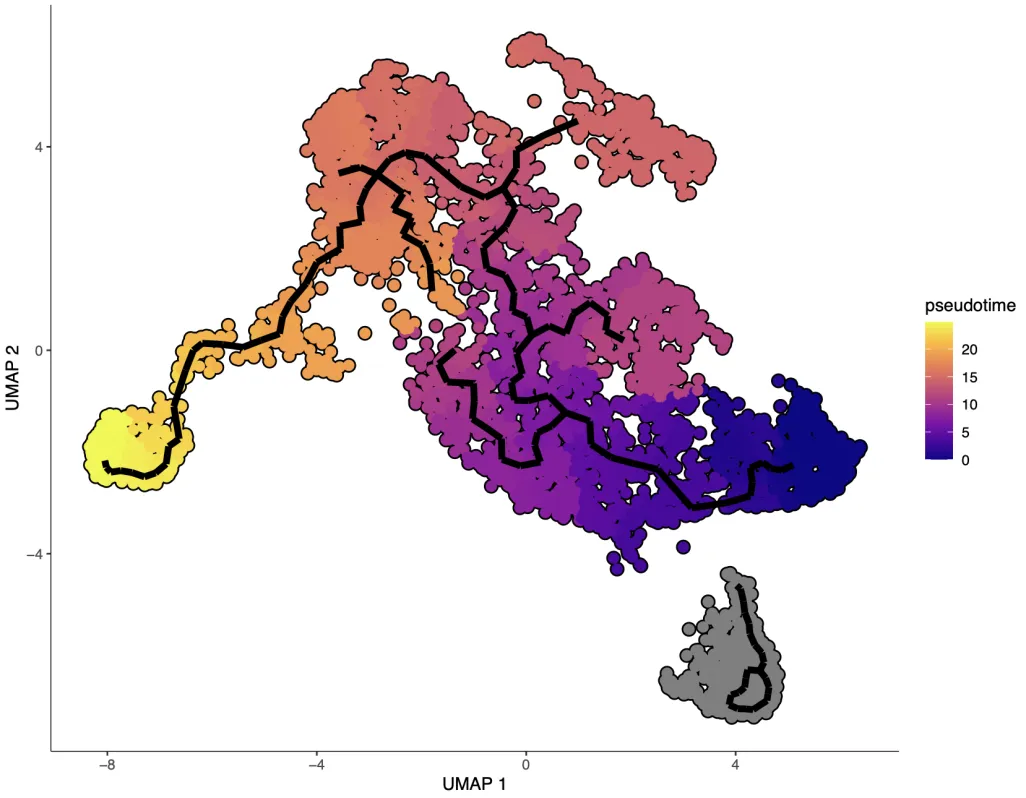
2D可视化
## 2D
plot_cells(cds, label_cell_groups = F,
color_cells_by = "pseudotime",
label_branch_points = F,
label_roots =F, # 显示根
label_leaves =F,
graph_label_size = 0,
cell_size=2,
trajectory_graph_color='black',
trajectory_graph_segment_size = 2)
ggsave("cds_trajectory.pdf",width = 9,height = 7)
qsave(cds,"cds_res.qs")
设定了起点之后进行图片可视化,但似乎轨迹没有很完美,最终没有落到CytoTRACE2中得到的分值最低的Treg细胞中。
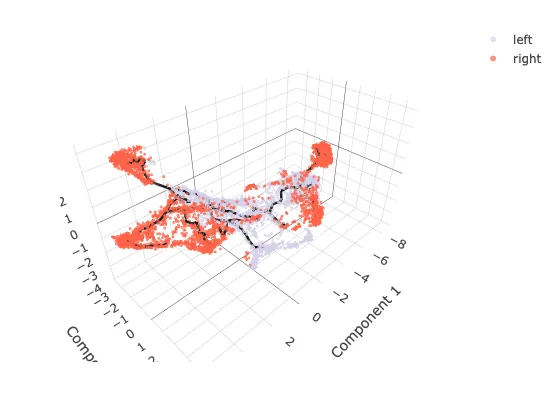
3D可视化
## 3D
cds_3d <- reduce_dimension(cds, max_components = 3)
cds_3d <- cluster_cells(cds_3d)
cds_3d <- learn_graph(cds_3d)
cds_3d <- order_cells(cds_3d, root_pr_nodes=get_earliest_principal_node(cds))
cds_3d_plot_obj <- plot_cells_3d(cds_3d,cell_size=5,color_palette = my60colors,
color_cells_by="location") # celltype
cds_3d_plot_obj
笔者设定了celltype之后没有显示颜色,也没找到解决办法,无奈之下采用location做替代展示。
9.轨迹差异基因分析
这里还可以做差异基因分析,图片就不展示了。
# 提取不同轨迹的差异基因/并选择前12个
# neighbor_graph="principal_graph"提取轨迹上相似位置是否有相关的表达
trace_genes <- graph_test(cds,
neighbor_graph = "principal_graph",
cores = 4)
track_genes_sig <- trace_genes %>%
top_n(n=12, morans_I) %>%
pull(gene_short_name) %>%
as.character()
# 差异基因绘制######################################
levels(Idents(scRNA)) #打出来细胞类型供复制
# [1] "ZSCAN12+T" "Naive T" "Th1" "Tm" "ZNF793+T" "ELK4+T" "Th17"
# [8] "Treg"
lineage_cds <- cds[rowData(cds)$gene_short_name %in% track_genes_sig,
colData(cds)$celltype %in% c("ZSCAN12+T",
"Naive T",
"Th1",
"Tm",
"ZNF793+T",
"ELK4+T",
"Th17",
"Treg")]
#lineage_cds <- order_cells(lineage_cds)
plot_genes_in_pseudotime(lineage_cds,
color_cells_by="celltype",
min_expr=0.5)
ggsave("degs_monocles.pdf",width = 8,height = 14)
# 细胞映射
plot_cells(cds, genes= track_genes_sig,
cell_size=1,
show_trajectory_graph=FALSE,
label_cell_groups=FALSE,
label_leaves=FALSE)
ggsave("track_genes_sig.pdf",width = 12,height = 9)
monocl3中目前弃用了热图绘制,笔者提供一种讨巧的办法,使用老俊俊的R包
但是这种方法绘制的热图,未必符合研究的生物学逻辑,请谨慎使用!
# laojunjun热图##################################
library(ClusterGVis)
genes <- row.names(subset(trace_genes, q_value < 0.01 & morans_I > 0.25))
mat <- pre_pseudotime_matrix(cds_obj = cds,gene_list = genes)
mat <- as.data.frame(mat)
head(mat[1:5,1:5])
# kmeans聚类
ck <- ClusterGVis::clusterData(obj = mat,
cluster.method = "kmeans",
cluster.num = 8)
# 富集分析
library(org.Hs.eg.db)
enrich <- enrichCluster(object = ck,
OrgDb = org.Hs.eg.db,
type = "BP", # 可以自己定义
pvalueCutoff = 0.05,
topn = 5, # topn设置为NULL可获得全部结果
seed = 123)
# 添加行注释
pdf('monocle3.pdf',height = 10,width = 12,onefile = F)
visCluster(object = ck,
plot.type = "both",
add.sampleanno = F,
markGenes.side = "left",
markGenes = sample(rownames(mat),30,replace = F),
ht.col.list = list(col_range = c(-2,-1.5,-1,-0.5,0,0.5,1,1.5,2),
col_color = c("#0D0887FF","#42049EFF","#6A00A8FF",
"#900DA4FF","white","#E16462FF",
"#FCA636FF","#FCCE25FF","#F0F921FF")),
genes.gp = c('italic',fontsize = 12,col = "black"),
line.side = "left",
annoTerm.data = enrich,
)
dev.off()
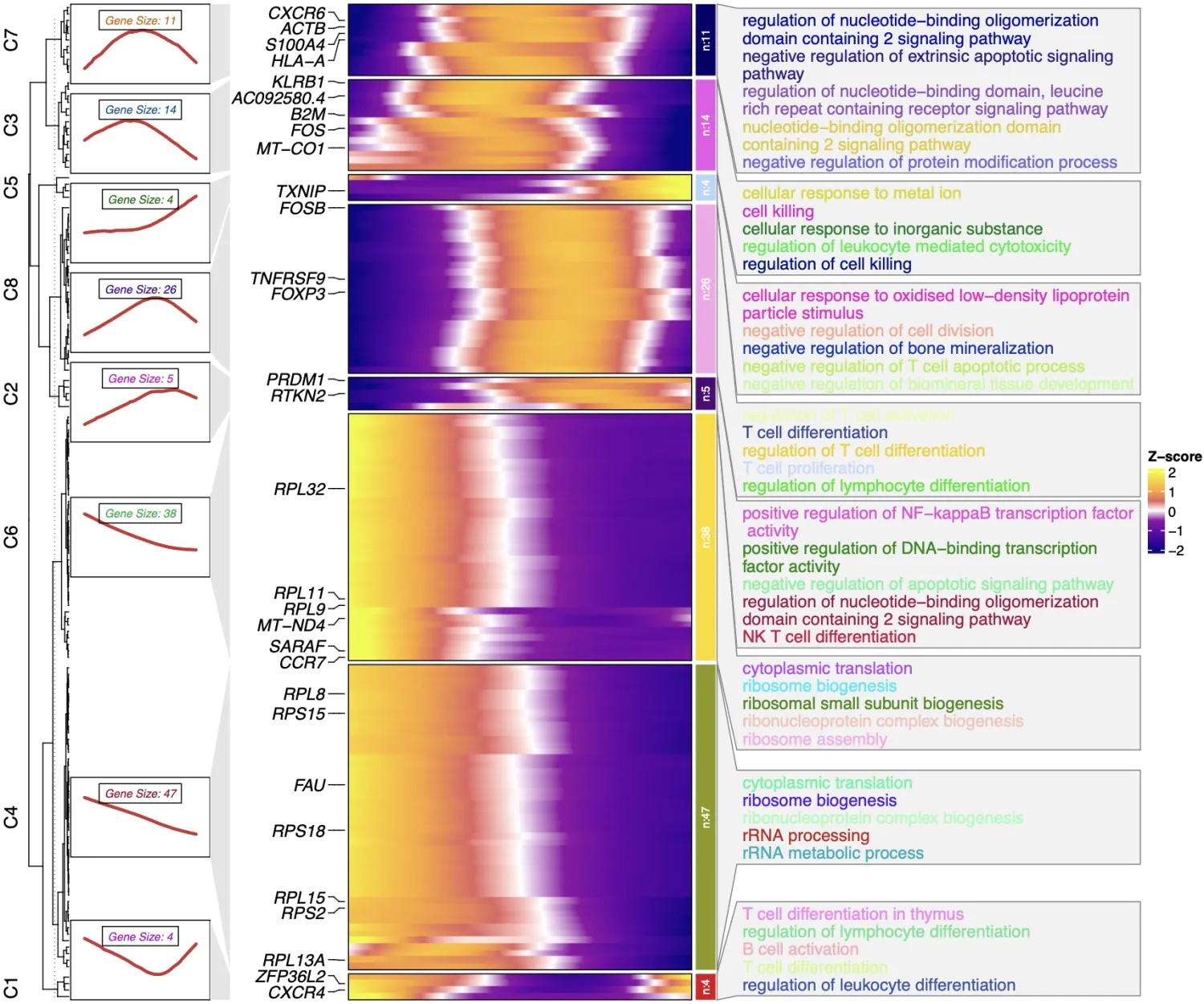
本次分析完成了cytoTRACE2和monocle3联用的实践流程。首先通过cytoTRACE2解析细胞分化轨迹起点,定位发育起源的起始细胞群体;继而运用monocle3建立拟时序模型,揭示不同细胞亚群在分化路径中的动态基因表达特征;基于获得的差异表达基因集,可进一步开展功能富集分析挖掘关键调控通路,最终通过生物学意义解读验证研究者提出的科学假说,当然本流程实验也主要是技术实操,并不考虑生物学逻辑。
参考资料:
-
Mapping single-cell developmental potential in health and disease with interpretable deep learning. 2024 Mar 21:2024.03.19.585637
-
The single cell transcriptional landscape of mammalian organogenesis. Nature. 2019 Feb;566(7745):496-502.
-
Reversed graph embedding resolves complex single-cell trajectories. Nat Methods. 2017 Oct;14(10):979-982.
-
CytoTRACE2:https://github.com/digitalcytometry/cytotrace2
-
生信技能树:https://mp.weixin.qq.com/s/S1-ClJEtR0ro0sYnIF6iaw https://mp.weixin.qq.com/s/YHJXm17qPrxuCVsZivHHNw https://mp.weixin.qq.com/s/Q4Vp-bSzLnomggeq-FaMRw https://mp.weixin.qq.com/s/XVG7rXMtq37EW8CsTmwgag https://mp.weixin.qq.com/s/fjZ2rLHB-D64rShOwWxNQQ
-
单细胞天地:https://mp.weixin.qq.com/s/wzn5Jdf5DR1LrhXecYg7Yw https://mp.weixin.qq.com/s/rgT5VCT0fqvhrTwZOtnaWA
-
生信星球:https://mp.weixin.qq.com/s/WvisYkVPCVQEfC4LKcgq2g https://mp.weixin.qq.com/s/qBJmwlGcwWCImm8lRkaPEA
-
老俊俊的生信笔记:https://mp.weixin.qq.com/s/7sYTrHlhnHj5490RyrdBug
-
生信方舟: https://mp.weixin.qq.com/s/aVUpRIkDi83B8_Y_BSBkVA https://mp.weixin.qq.com/s/NRrFH8sjdUUq20z9hWAFyQ https://mp.weixin.qq.com/s/vnTh5hVoKzGBjjCU1fmaYQ
-
KS科研分享与服务:https://mp.weixin.qq.com/s/Cyng4LjHihmjajr1pT0v1A
-
Trajectory inference analysis of scRNA-seq data: https://www.youtube.com/watch?v=XmHDexCtjyw&list=PLjiXAZO27elC_xnk7gVNM85I2IQl5BEJN&index=12
致谢:感谢曾老师以及生信技能树团队全体成员。更多精彩内容可关注公众号:生信技能树,单细胞天地,生信菜鸟团等公众号。
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多相关内容可关注公众号:生信方舟
- END -


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









