







在下载完数据之后就需要对数据进行后续的分析,既往推文可见 单细胞上游分析/cellranger流程学习(一):https://mp.weixin.qq.com/s/6l2jrb2jMzqgJp01-7yvlQ
此外再回顾一下CNCB中的数据下载方式,并选择一个比较复杂的数据实操练习一下, CNCB(国家生物信息中心)数据下载流程学习:https://mp.weixin.qq.com/s/h5OxkU7WKePYbl53hH8sMQ
数据下载:
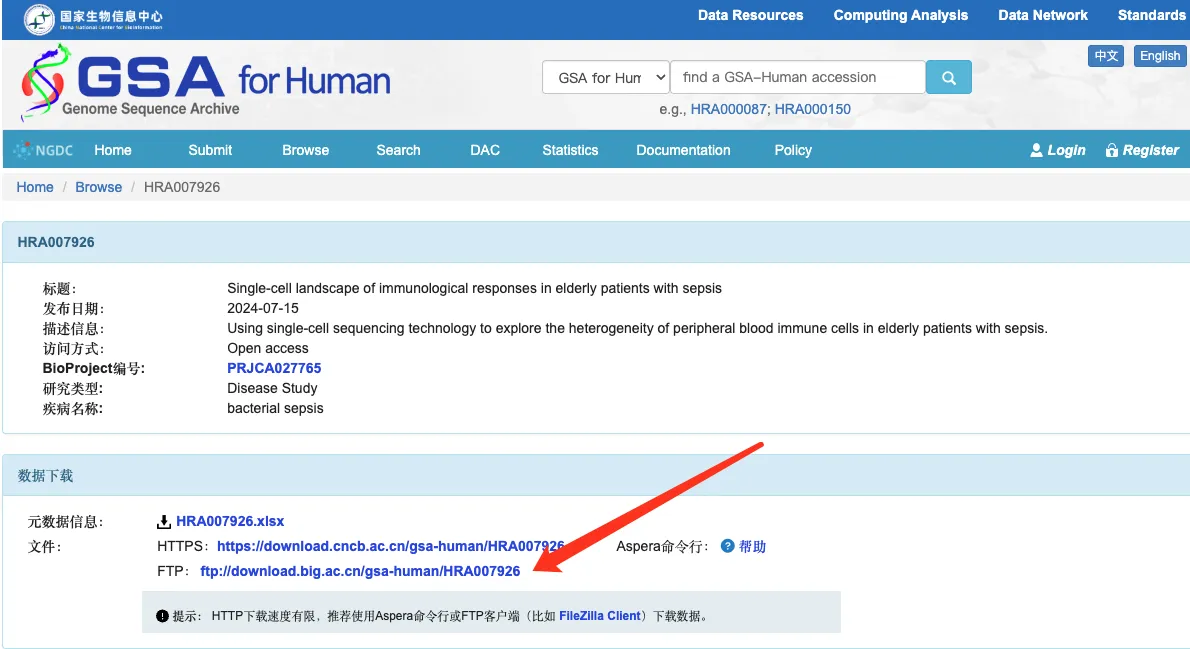
wget -r -nH --cut-dirs=2 --ftp-user=anonymous --ftp-password=anonymous ftp://download.big.ac.cn/gsa-human/HRA007926/ -P /home/data/t200558/scRNAdata/HRA007926
# 后台运行
nohup wget -c -r -nH --cut-dirs=2 --ftp-user=anonymous --ftp-password=anonymous ftp://download.big.ac.cn/gsa-human/HRA007926/ -P /home/data/t200558/scRNAdata/HRA007926 > download.log 2>&1 &
-
nohup:这个命令的作用是使后续的命令在当前用户退出或关闭终端时依然可以继续运行。nohup 会忽略挂起信号并让进程继续执行。
-
wget:wget 是一个常用的工具,用于从网络上下载文件或内容,支持 HTTP、HTTPS 和 FTP 等协议。
-
-c:继续下载。这个选项允许你从上次下载中断的地方继续下载,而不是从头开始。
-
-r:递归下载。它告诉 wget 下载整个目录结构中的所有文件,而不仅仅是单个文件。
-
-nH:不创建主机目录(nH 代表 "no host directories"),通常 wget 会在下载文件时以网站的主机名作为目录名来组织文件,这个选项会禁用这一行为。
-
--cut-dirs=2:用于修改保存文件时的目录结构。--cut-dirs=2 表示从服务器的路径中剔除前两个目录,并将剩下的目录结构保存在本地。这个选项有助于简化保存的文件路径。
-
--ftp-user=anonymous --ftp-password=anonymous:提供 FTP 服务器的用户名和密码。在这里,使用的是匿名用户(anonymous)。
-
ftp://download.big.ac.cn/gsa-human/HRA007926/:这是待下载的 FTP 服务器地址和文件路径。
-
-P /home/data/t200558/scRNAdata/HRA007926:指定下载的文件保存目录。在这里,所有下载的文件会保存到 /home/data/t200558/scRNAdata/HRA007926 目录中。
-
download.log 2>&1:将标准输出(stdout)和标准错误输出(stderr)都重定向到 download.log 文件中。这样可以记录下载过程中的所有输出(包括错误信息)。
-
&:将命令放入后台运行。这样可以在终端中继续进行其他操作,而不必等到下载完成。
数据检查
下载完数据之后需要仔细核对一下数量以及跟官方信息对应情况
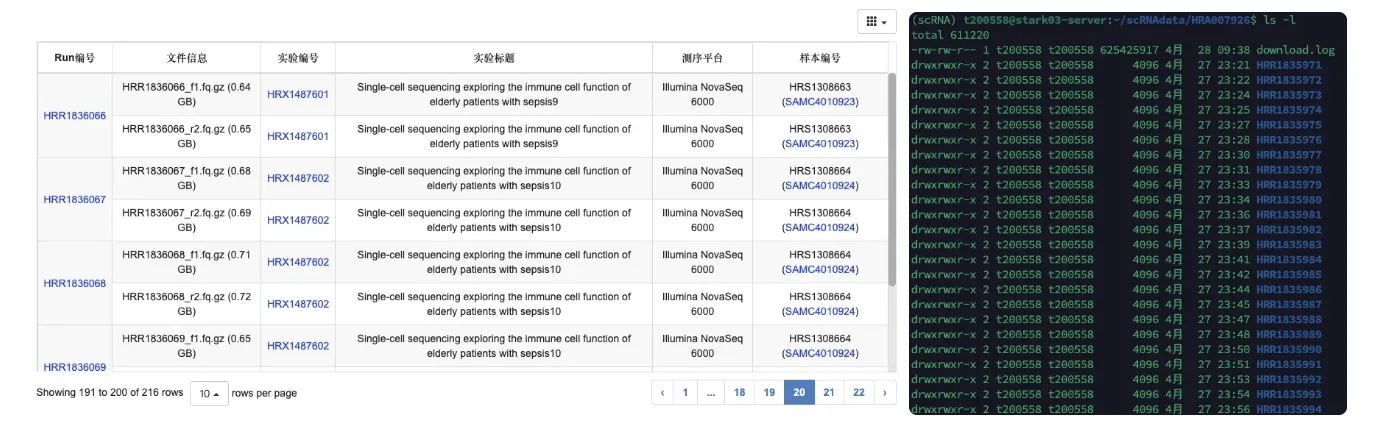
# 文件夹是以d开头的,可以通过下面代码组合进行计算
ls -l | grep ^d | wc -l
# 108 ,可发现108个文件夹,因此数量上是正确的
# 也可以计算所有文件和文件夹的数量
ls -l | wc -l
数据改名
该数据改名比较麻烦,首先是不同样本有很多个Run编号样本,并且还存在于不同的文件夹中,那么为了避免后续混淆。笔者首先按照不同文件夹把里面的文件按照规范命名,然后再把规范命名的文件按照相同样本汇总到一起后再进行样本名修改,最后把所有完成命名的文件放到一起进行后续分析。
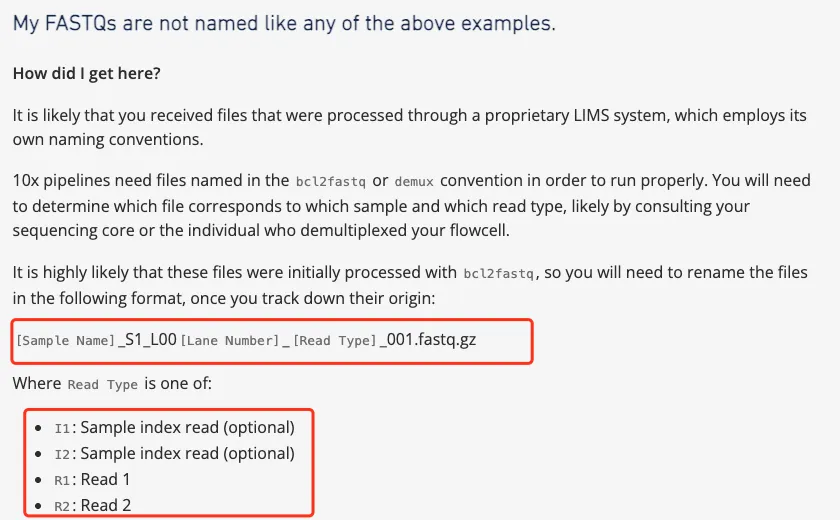
1.按照不同文件夹把里面的文件按照规范命名
其实也可以先不规范命名后面再命名
# 遍历所有文件夹
for dir in */; do
# 获取文件夹的名字,作为样本名
sample_name=$(basename "$dir")
# 进入当前文件夹
cd "$dir"
# 查找所有文件,并按10X的命名规则重命名
for file in *.fq.gz; do
if [[ "$file" =~ _f1.fq.gz ]]; then
# 重命名R1文件
mv "$file" "${sample_name}_S1_L001_R1_001.fastq.gz"
elif [[ "$file" =~ _r2.fq.gz ]]; then
# 重命名R2文件
mv "$file" "${sample_name}_S1_L001_R2_001.fastq.gz"
fi
done
# 返回上一层目录
cd ..
done
检查是否成功
find . -print
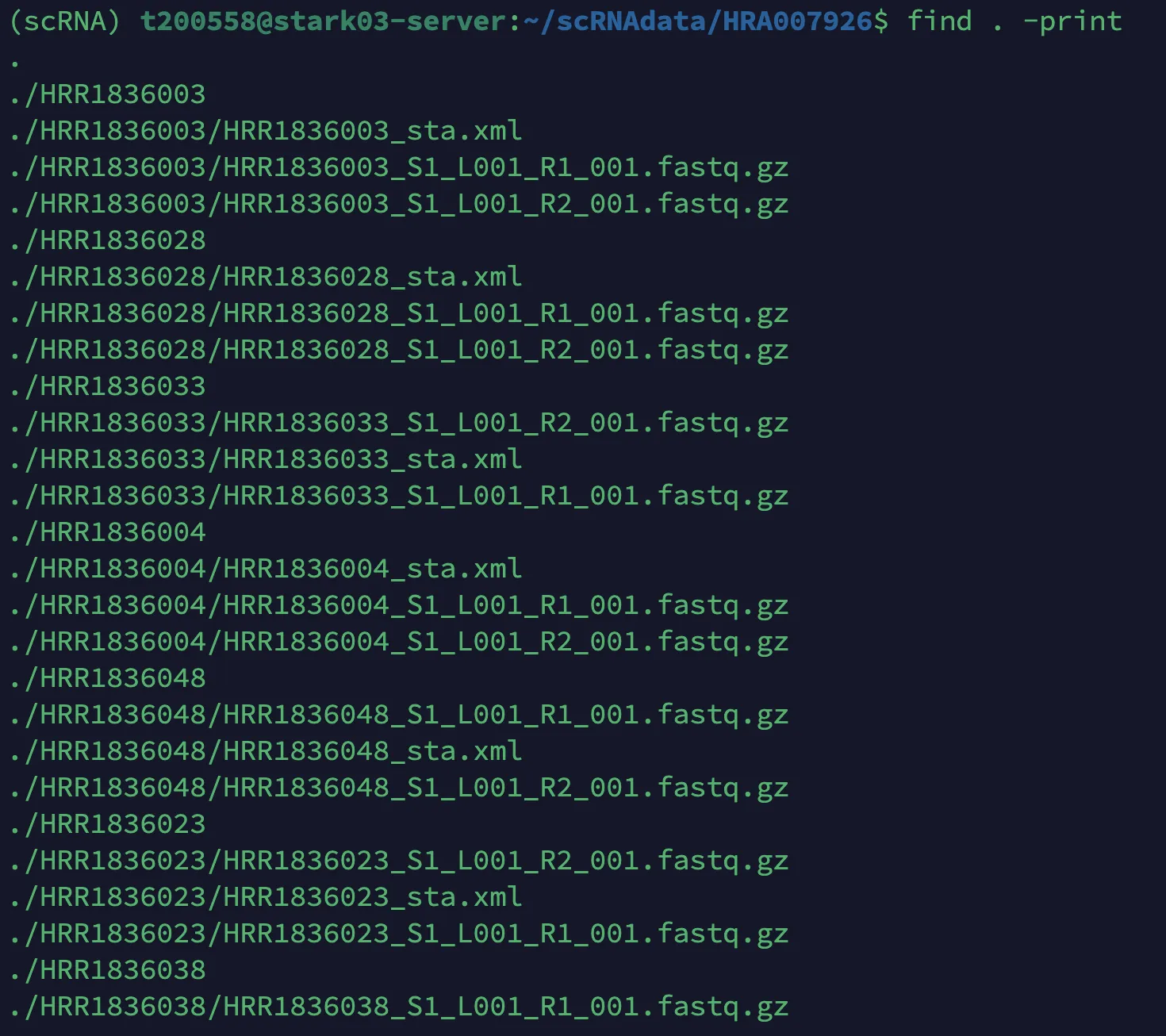
2.规范命名的文件按照样本汇总到新的文件夹中并按照样本重新命名
不同人有不同的习惯,笔者喜欢对着官方信息检查数据
# 创建以样本编号命名的文件夹
mkdir -p HRS1308655
# 移动属于该样本的文件进入该文件夹
# 要非常仔细检查样本
mv HRR183597{1..4}/*.fastq.gz HRS1308655/
# 检查文件
ls -l HRS1308655/
# 其他的文件也按照这个流程进行移动
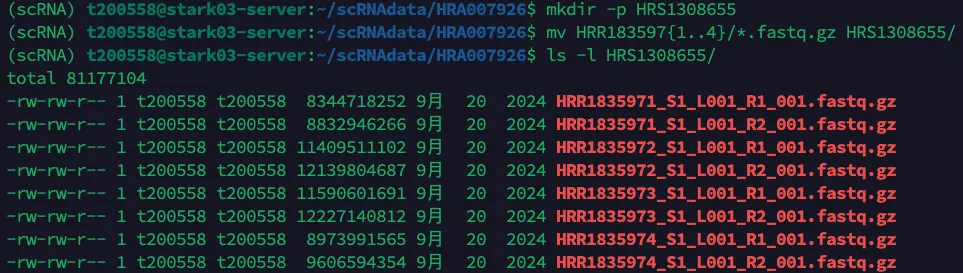
然后再将该文件夹中的文件进行重命名,需要把不同的Run编号(HRR183597{1..4})修改成相同的样本名称,但是S1需要按照不同的Run编号数量进行修改,因此最后格式需要改成:[sample name]_S[1-4]_L001_R1/2_001.fastq.gz,其中S可以理解为segment,然后把不同编号的segment汇总在一起。如果嫌弃改名称很麻烦,其实可以手动修改的,修改名称代码一定要借助大模型,千万别自己死磕..
# 获取当前文件夹名称(例如 HRS1308664)
folder_name=$(basename "$PWD")
# 提取所有唯一的 HRR 前缀,并生成 S1/S2/S3... 的映射
declare -A hr_mapping
counter=1
for file in HRR*.fastq.gz; do
# 提取 HRR 前缀(例如 HRR1836007)
hr_prefix=$(echo "$file" | grep -oE '^HRR[0-9]+' | head -1)
# 去重并分配 S 编号
if [ -n "$hr_prefix" ] && [ -z "${hr_mapping[$hr_prefix]}" ]; then
hr_mapping["$hr_prefix"]="S$counter"
((counter++))
fi
done
# 重命名文件
for file in HRR*.fastq.gz; do
# 分解文件名各部分
hr_prefix=$(echo "$file" | grep -oE '^HRR[0-9]+' | head -1)
s_id="${hr_mapping[$hr_prefix]}" # 获取新 S 编号(如 S1)
# 提取剩余部分,并移除原 S1 字段(第一个下划线后的内容)
rest_part=$(echo "$file" | sed "s/^${hr_prefix}_//")
rest_part_modified=$(echo "$rest_part" | cut -d'_' -f2-) # 去掉原 S1
# 构建新文件名(格式:HRS1308664_S1_L001_R1_001.fastq.gz)
new_name="${folder_name}_${s_id}_${rest_part_modified}"
# 执行重命名(安全模式,避免覆盖)
mv -nv "$file" "$new_name"
done
3.移动文件到新的文件夹中做最后工作准备
# 创建新文件夹
mkdir merged_HRS
# 移动文件到该文件夹中
find HR* -type f -name "*.fastq.gz" -exec mv -v {} merged_HRS/ \;
# 查看所有以fastq.gz结尾的文件,并统计数量
find merged_HRS -type f -name "*.fastq.gz" | wc -l
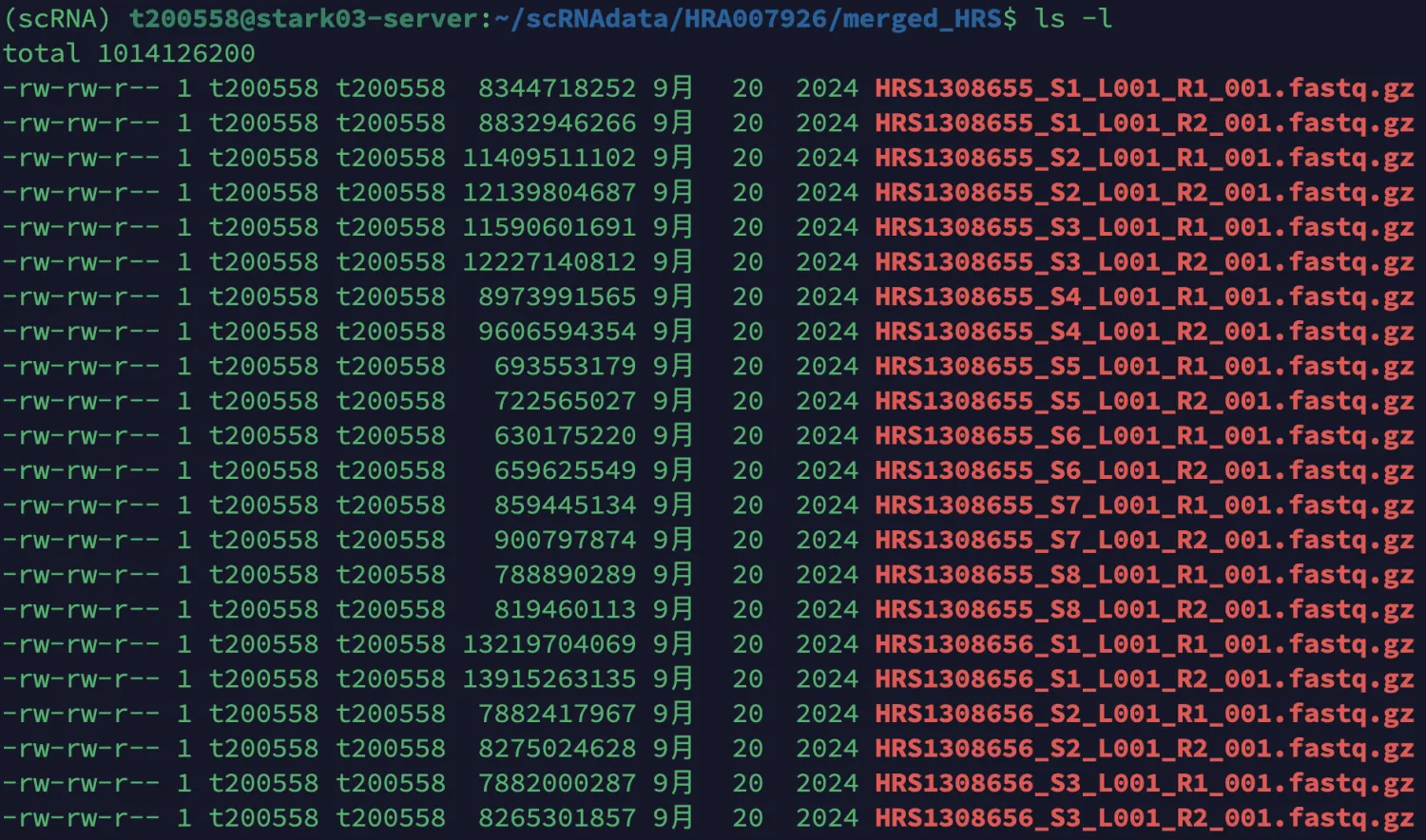
Cellranger运行
# 删除当前目录下(不递归子目录)的 HRS/HRR 开头文件夹
# 删之前一定要谨慎哈!!
rm -rf HRS*/ HRR*/
# 临时添加到PATH
export PATH=/home/data/t200558/biosoft/cellranger-9.0.0/bin:$PATH
# which函数确认是否添加成功
which cellranger
# 获得所有sample名称,进入merged_HRS文件夹提取
ls *.fastq.gz | cut -d'_' -f1 | sort -u > ../samples.txt
# 设定参考基因组
ref="/home/data/t200558/reference/GRCh38/refdata-gex-GRCh38-2024-A"
# 设定fastq文件路径
fastqs="/home/data/t200558/scRNAdata/HRA007926/merged_HRS"
# 输出文件存放路径
output="/home/data/t200558/scRNAdata/HRA007926"
# Cellranger 后台运行(每个样本独立日志)
while read hrs_id; do
nohup cellranger count \
--id="$hrs_id" \
--transcriptome="$ref" \
--fastqs="$fastqs" \
--sample="$hrs_id" \
--create-bam=true \
--nosecondary \
--localcores=4 \
--localmem=30 \
> "${hrs_id}.log" 2>&1 &
done < samples.txt
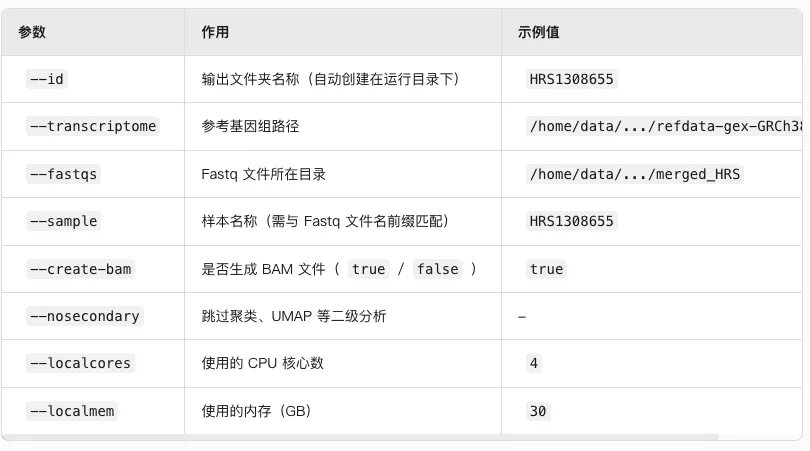
关于参数Cellranger中的参数解析,让大模型试着整理了一下便于查看。同时进行后台运行并对每一个文件都生成一个运行日志以便于查看。
致谢:感谢含盐量过高的鱼提供了部分指导~
参考资料:
-
10X genomics-cellranger:https://www.10xgenomics.com/support/software/cell-ranger/latest/tutorials/cr-tutorial-in
-
10X Genomics 帮助手册:https://assets.ctfassets.net/an68im79xiti/1CnKSfa7taoQwIEe0WaA4m/8635b2c9ee86c022e731b6fb2e13fed2/CG000080_10x_Technical_Note_Base_Composition_SC3_v2_RevB.pdf
-
生信技能树:https://mp.weixin.qq.com/s/X1SYGr2zv-b7hSppNC_22Q https://mp.weixin.qq.com/s/X1SYGr2zv-b7hSppNC_22Q
-
生信菜鸟团:https://mp.weixin.qq.com/s/lvooimNKTQ8U9_2t2SuweQ https://mp.weixin.qq.com/s/AVqv07swFvjl6OCnLwwLPA
-
单细胞天地:https://mp.weixin.qq.com/s/XAn89z-pPRcPExEQ8IkhpQ
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多相关内容可关注公众号:生信方舟
- END -


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









