







在上一步对数据进行Cellranger count分析之后,接下来可以简单看一下每个样本得到的结果文件和Summary内容,既往推文可见 单细胞上游分析/cellranger流程学习(二):https://mp.weixin.qq.com/s/649AtykVmBRz2IjlRH9MUQ
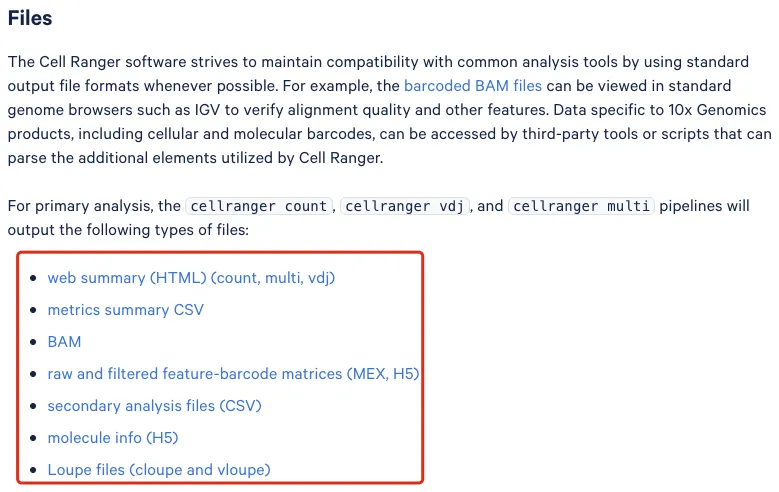
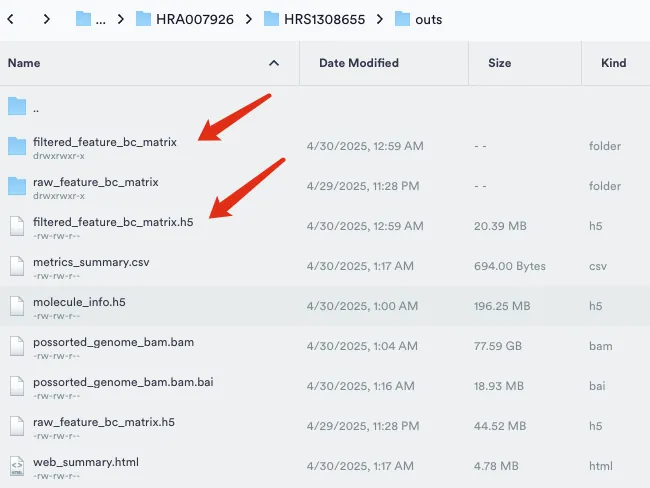
在分析结束的时候最主要关注"outs"这个文件夹,里面包含了后续所需要用到的文件。官网信息也告诉了我们针对cellranger count,cellranger vdj 和 cellranger multi分析之后最终会得到以下的文件:
-
Web summary(HTML)(count, multi,vdj): 一个交互式网页报告,总结了数据的整体质量,包括了测序质量、reads mapping信息、barcode分布、UMI数量、细胞数估计、基因数等核心指标。
-
Metrics summary.csv:是Web summary.html中统计指标的表格版本;
-
BAM:reads比对到参考基因组后的二进制文件,包括read序列,位置,CB(cell barcode),UB(UMI barcode)等信息;
-
Raw and Filtered Feature-Barcode Matrices (MEX, H5):raw文件所有检测到的 barcode,包括空滴和低质量细胞。filtered文件经过 Cell Ranger 细胞识别算法筛选出来的“真实”细胞。
-
Secondary Analysis Files (CSV):包括 PCA、t-SNE、聚类、差异表达等结果的 CSV 表格。是 Cell Ranger 自动完成的基本分析结果。
-
Molecule Info (H5):是包含每个 reads 的详细信息(Unique Molecular Identifier(UMI,唯一分子标识符)、barcode、基因等)的原始数据。用于重做某些分析(比如重计算 UMI 聚合、RNA velocity、RNA-seq 重聚类等)。
以HRS1308655样本为例,查看其Web summary(HTML)结果
Summary结果
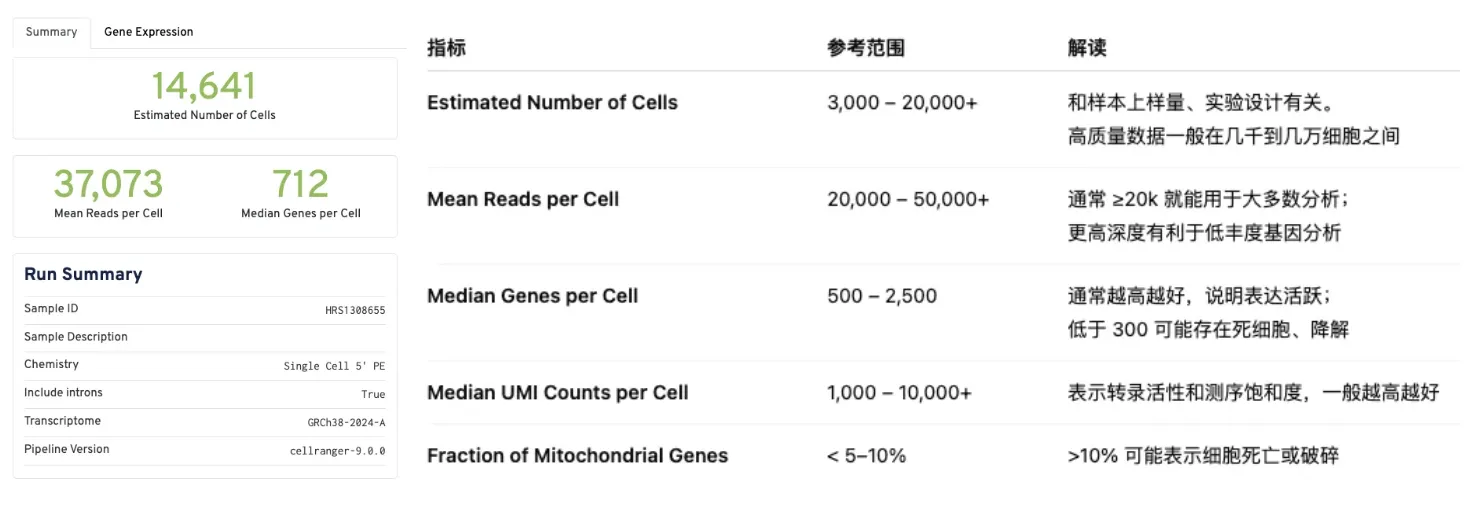
估计细胞数:14641个,每个细胞平均读数:37073条,每个细胞的中位基因数:712个;其他包括了样本ID,样本描述,化学方法,是否含有内含子,参考转录组信息,Cellranger版本。以下指标的正常范围仅供参考。
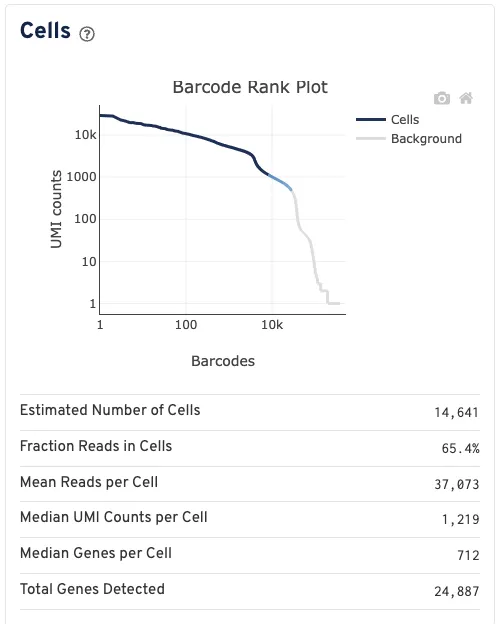
细胞数目评估信息
-
估计细胞数 (Estimated Number of Cells):与至少一个细胞相关联的条形码数量。
-
细胞中读数的比例 (Fraction Reads in Cells):具有有效条形码且被高置信度比对到基因组,并关联到细胞条形码的读数占比。
-
每个细胞的平均读数 (Mean Reads per Cell):测序总读数除以与含细胞分区关联的条形码数量。
-
每个细胞的中位UMI计数 (Median UMI Counts per Cell):每个细胞关联条形码的UMI计数中位值。
-
每个细胞的中位基因数 (Median Genes per Cell): 每个细胞关联条形码检测到的基因数中位值(定义为至少存在1个UMI计数)。
-
检测到的总基因数 (Total Genes Detected): 在任何细胞中至少存在1个UMI计数的基因数量。
-
条形码排名图 (Barcode Rank Plot): 该图显示映射到每个条形码的过滤后UMI计数。条形码是否被判定为细胞关联不严格基于UMI计数,而可能根据其表达谱确定,或通过蛋白质聚集体检测过滤和/或高占用率Gel Bead-In Emulsion(GEM, 乳滴凝胶珠)过滤移除。因此,图中某些区域可能同时包含细胞关联和背景关联的条形码。这些区域的颜色基于局部细胞关联条形码的密度。将鼠标悬停在图表上会显示该区域中被判定为细胞的条形码总数及占比,以及这些条形码的UMI计数和按UMI计数降序排列的条形码排名。如果这个曲线出现一个明显徒降的趋势,这表明与细胞相关的barcode和空白的barcode区分的很好。

其中Low Fraction Reads in Cells的比率理想情况应该是>70%,但实际上如果数值没有相差很大还是会接着进行后续分析。
测序和比对信息

一、测序部分
-
Number of Reads(总读数):分配给该文库的测序读长对总数:542,784,604。
-
Number of Short Reads Skipped(跳过的短读数):因不满足最小长度要求而被过滤的读长对数量:0。
-
Valid Barcodes(有效条形码比例):经过校正后与白名单匹配的条形码读长占比:88.4%。
-
Valid UMIs(有效UMI比例):不含N碱基且非均聚物的UMI读长占比:99.9%。
-
Sequencing Saturation(测序饱和度):来自已观测到UMI的读长占比,反映文库复杂性和测序深度:84.3%。
-
Q30 Bases in Barcode(条形码中Q30碱基比例):基于barcode的质量分数,大于30的比率:94.6%。
-
Q30 Bases in RNA Read(RNA读长中Q30碱基比例):基于RNA reads的质量分数,大于30的比率:86.9%。
-
Q30 Bases in RNA Read 2(第二条RNA读长中Q30碱基比例):质量分数≥30的RNA读长2碱基占比:81.5%。
-
Q30 Bases in UMI(UMI中Q30碱基比例):基于UMI的质量分数,大于30的比率:93.6%。
二、比对部分
-
Reads Mapped to Genome(映射到基因组的读数比例):比对到基因组的读长总占比(含多位置比对):85.9%。
-
Reads Mapped Confidently to Genome(可靠映射到基因组的读数比例):唯一比对到基因组的读长占比:75.7%。
-
Reads Mapped Confidently to Intergenic Regions(可靠映射到基因间区的读数比例):唯一比对到基因间非编码区域的读长占比:8.8%。
-
Reads Mapped Confidently to Intronic Regions(可靠映射到内含子区的读数比例):唯一比对到基因内含子区域的读长占比:5.6%。
-
Reads Mapped Confidently to Exonic Regions(可靠映射到外显子区的读数比例):唯一比对到基因外显子区域的读长占比:61.4%。
-
Reads Mapped Confidently to Transcriptome(可靠映射到转录组的读数比例):唯一比对到注释转录本(不含内含子)的读长占比:56.5%。
-
Reads Mapped Antisense to Gene(反义链映射的读数比例):比对到基因反义链且无正义链比对的读长占比:9.9%。
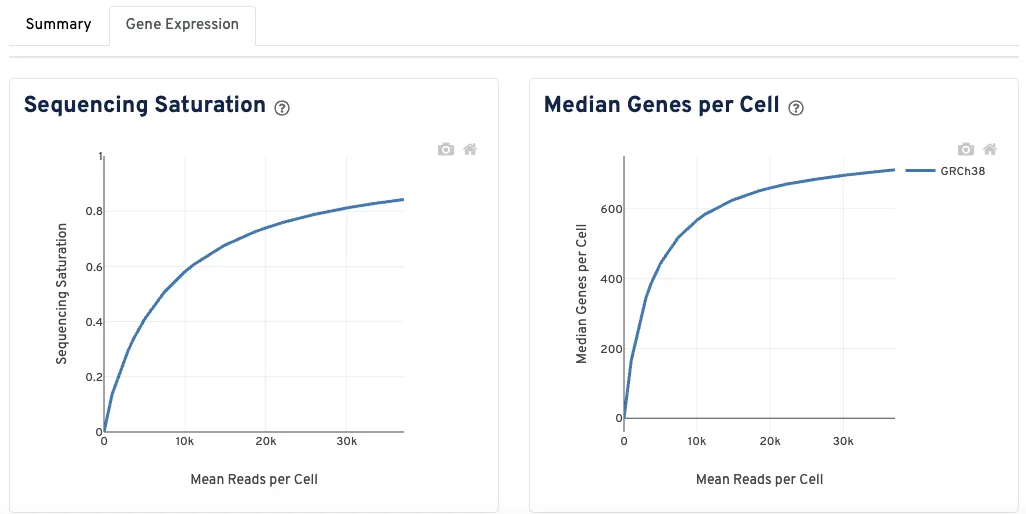
基因表达情况
-
左图展示了测序饱和度指标随采样测序深度(以每个细胞平均读数为单位)的变化情况,最大深度为实际观测到的测序深度。测序饱和度是衡量观测到的文库复杂度的指标,当所有转换的mRNA转录本被测序时,该值趋近于1.0(100%)。
-
右图展示了每个细胞的中位基因数随采样测序深度(以每个细胞平均读数为单位)的变化情况,最大深度为实际观测到的测序深度。
-
曲线末端附近的斜率可解释为:超过当前测序深度后,继续增加测序深度所能获得的潜在收益上限,越平缓则说明越没有作用了。
最后使用过滤后文件即可进行分析。
整套流程运行速度较慢,尤其生成BAM文件的步骤不仅占用大量存储空间,还显著增加了算力消耗。再感慨一下,计算机资源同样是珍贵且有限的资源..
参考资料:
-
Cellranger 10X 官方:https://www.10xgenomics.com/support/software/cell-ranger/latest/analysis/outputs/cr-outputs-web-summary-count
-
单细胞天地:https://mp.weixin.qq.com/s/yHWoJOGJnndV158a7AVFPA
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多相关内容可关注公众号:生信方舟
- END -

















 3226
3226

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









