







blast
基本介绍
BLAST (Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。BLAST程序能迅速与公开数据库进行相似性序列比较。BLAST结果中的得分是对一种对相似性的统计说明。
- blastp是蛋白序列到蛋白库中的一种查询。库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
- blastx是核酸序列到蛋白库中的一种查询。先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。
- blastn是核酸序列到核酸库中的一种查询。库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。
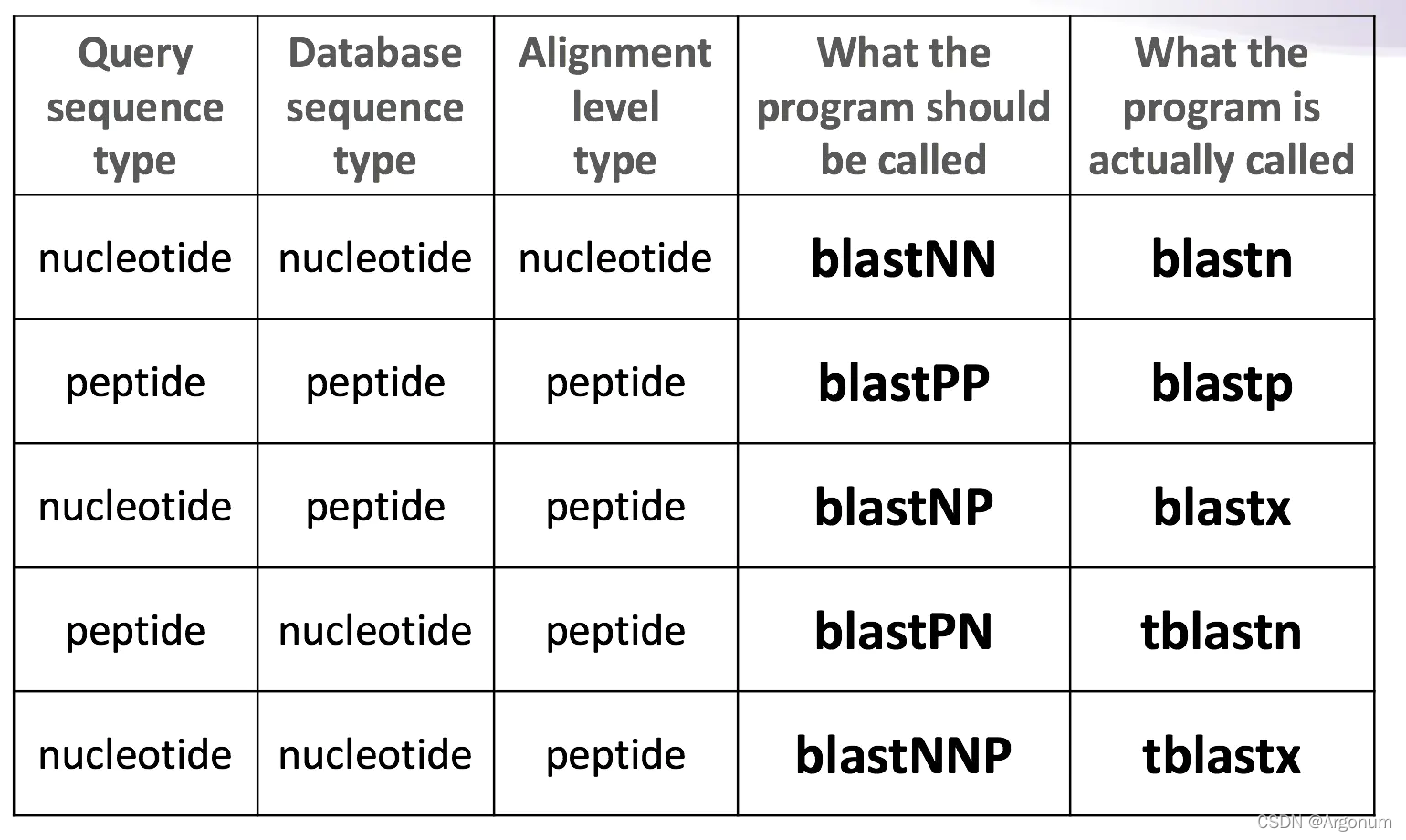
参数介绍
博客来源https://www.jianshu.com/p/de28be1a3bea
-
-db : 数据库在本地的位置,或者是NCBI上数据库的类型
-
-query: 检索文件
-
-query_loc : 指定检索的位置
-
-strand: 搜索正义链还是反义链,还是都要
-
out : 输出文件
-
-remote: 可以用NCBI的远程数据库, 一般与 -db nr
-
-evalue 科学计数法,比如说1e3,定义期望值阈值。E值表明在随机的情况下,其它序列与目标序列相似度要大于这条显示的序列的可能性。 与S值有关,S值表示两序列的同源性,分值越高表明它们之间相似的程度越大 E值总结:
1.E值适合于有一定长度,而且复杂度不能太低的序列。2. 当E值小于10-5时,表明两序列有较高的同源性,而不是因为计算错误。3. 当E值小于10-6时,表时两序列的同源性非常高,几乎没有必要再做确认。 -
-gapopen,打开gap的代价;
-
-gapextend, gap延伸的代价;
-
-penalty:核酸错配的惩罚;
-
-reward, 核酸正确匹配的奖励;
-
结果过滤:-perc_identity, 根据相似度
busco
基本介绍
参考这篇博客吧,巨详细,贼好用
https://www.jianshu.com/p/5041460f7a5d
基本介绍
原文链接https://blog.csdn.net/u010608296/article/details/101713130
wgsim是一块用于高通量数据模拟的软件,whole genome simulation。这款软件可以模拟出illumina测序数据,并且可以自由调整测序reads的读长,插入片段大小以及错误率等,使用起来比较方便。模拟数据主要用于软件的测试与评估。例如对序列拼接软件的评估。因为模拟数据是根据已有的参考序列来的,我们可以将模拟出来的数据进行拼接,再将拼接的结果与原序列进行比对。这样就能每次调整单因素变量,例如比较不同reads读长,不同插入片段大小或者不同错误率条件下,对序列拼接的影响。
也可以为参考序列模拟变异位点,例如点突变、片段获得缺失等,然后模拟数据,评估软件是否可以检测出这些变异位点。
- -e 是错误率,默认是0.02
- -d reads两头的距离,也就是插入片段长度,默认250bp,注意插入片段本身是包含reads长度的,而不是reads之间的距离
- -s 是-d插入片段的偏差,默认是20,也就是-d的值加减20,我们知道插入片段长度并不是固定的,而是一个范围
- -N 是测序的层数,控制输出数据量
- -1 是reads1长度,默认70bp
- -2 是reads2长度,默认70bp
- -r 突变率
- -R -X 都是调整indels的
- -h 是单倍体模式

















 518
518

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









