







Amber20 蛋白质-小分子分子动力学模拟
一、体系的准备
将蛋白质-小分子复合物的 pdb 文件放在工作目录下(比如之前使用 AutoDock 进行分子对接得到的蛋白质-小分子复合物)
username@computer:~/test$ ls
ligand MD original_complex.pdb receptor
username@computer:~/test$ vi original_complex.pdb

1.1 小分子的准备工作
1.1.1 从复合物中分离小分子
username@computer:~/test$ awk '$1=="HETATM"' original_complex.pdb | awk '$4=="UNL"' > ligand.pdb
username@computer:~/test$ ls
ligand ligand.pdb MD original_complex.pdb receptor
username@computer:~/test$ mv ligand.pdb /home/username/test/ligand
username@computer:~/test$ cd ligand
username@computer:~/test/ligand$ vi ligand.pdb
1.1.2 为小分子加氢
将上一步分离出的小分子用 Windows 系统下的 Pymol 软件进行加氢并命名为 ligand_h.pdb 后传输回服务器内。
username@computer:~/test/ligand$ ls
ligand_h.pdb ligand.pdb
1.1.3 为小分子重新编号
username@computer:~/test/ligand$ pdb4amber -i ligand_h.pdb -o ligand_h_renum.pdb
==================================================
Summary of pdb4amber for: ligand_h.pdb
===================================================
----------Chains
The following (original) chains have been found:
---------- Alternate Locations (Original Residues!))
The following residues had alternate locations:
None
-----------Non-standard-resnames
UNL
---------- Mising heavy atom(s)
None
1.1.4 生成小分子 mol2 文件
username@computer:~/test/ligand$ antechamber -fi pdb -fo mol2 -i ligand_h_renum.pdb -o ligand.mol2 -c bcc -pf y -nc 0
Welcome to antechamber 21.0: molecular input file processor.
acdoctor mode is on: check and diagnose problems in the input file.
The atom type is set to gaff; the options available to the -at flag are
gaff, gaff2, amber, bcc, and sybyl.
-- Check Format for pdb File --
Status: pass
-- Check Unusual Elements --
Status: pass
-- Check Open Valences --
Status: pass
-- Check Geometry --
for those bonded
for those not bonded
Status: pass
-- Check Weird Bonds --
Status: pass
-- Check Number of Units --
Status: pass
acdoctor mode has completed checking the input file.
Info: Total number of electrons: 184; net charge: 0
Running: /home/program/amber20/bin/sqm -O -i sqm.in -o sqm.out
1.1.5 生成小分子 frcmod 文件
username@computer:~/test/ligand$ parmchk2 -i ligand.mol2 -o ligand.frcmod -f mol2
1.1.6 生成小分子 lib 文件
username@computer:~/test/ligand$ vi tleap.in
username@computer:~/test/ligand$ tleap -f tleap.in
-I: Adding /home/program/amber20/dat/leap/prep to search path.
-I: Adding /home/program/amber20/dat/leap/lib to search path.
-I: Adding /home/program/amber20/dat/leap/parm to search path.
-I: Adding /home/program/amber20/dat/leap/cmd to search path.
-f: Source tleap.in.
Welcome to LEaP!
(no leaprc in search path)
Sourcing: ./tleap.in
----- Source: /home/program/amber20/dat/leap/cmd/leaprc.gaff2
----- Source of /home/program/amber20/dat/leap/cmd/leaprc.gaff2 done
Log file: ./leap.log
Loading parameters: /home/program/amber20/dat/leap/parm/gaff2.dat
Reading title:
AMBER General Force Field for organic molecules (Version 2.11, May 2016)
----- Source: /home/program/amber20/dat/leap/cmd/leaprc.protein.ff14SB
----- Source of /home/program/amber20/dat/leap/cmd/leaprc.protein.ff14SB done
Log file: ./leap.log
Loading parameters: /home/program/amber20/dat/leap/parm/parm10.dat
Reading title:
PARM99 + frcmod.ff99SB + frcmod.parmbsc0 + OL3 for RNA
Loading parameters: /home/program/amber20/dat/leap/parm/frcmod.ff14SB
Reading force field modification type file (frcmod)
Reading title:
ff14SB protein backbone and sidechain parameters
Loading library: /home/program/amber20/dat/leap/lib/amino12.lib
Loading library: /home/program/amber20/dat/leap/lib/aminoct12.lib
Loading library: /home/program/amber20/dat/leap/lib/aminont12.lib
Loading Mol2 file: ./ligand.mol2
Reading MOLECULE named UNL
Loading parameters: ./ligand.frcmod
Reading force field modification type file (frcmod)
Reading title:
Remark line goes here
Checking 'MOL'....
Checking parameters for unit 'MOL'.
Checking for bond parameters.
Checking for angle parameters.
Unit is OK.
Creating ligand.lib
Building topology.
Building atom parameters.
Checking Unit.
Building topology.
Building atom parameters.
Building bond parameters.
Building angle parameters.
Building proper torsion parameters.
Building improper torsion parameters.
total 13 improper torsions applied
Building H-Bond parameters.
Incorporating Non-Bonded adjustments.
Not Marking per-residue atom chain types.
Marking per-residue atom chain types.
(Residues lacking connect0/connect1 -
these don't have chain types marked:
res total affected
UNL 1
)
(no restraints)
Quit
Exiting LEaP: Errors = 0; Warnings = 0; Notes = 0.
1.2 蛋白质的准备工作
1.2.1 从复合物中分离蛋白质
username@computer:~/test$ awk '$1=="ATOM"' original_complex.pdb | awk '$5=="A"' > receptor.pdb
username@computer:~/test$ ls
ligand MD original_complex.pdb receptor receptor.pdb
username@computer:~/test$ mv receptor.pdb /home/username/test/receptor
username@computer:~/test$ cd receptor
username@computer:~/test/receptor$ vi receptor.pdb
1.2.2 为蛋白质进行修复
username@computer:~/test/receptor$ pdb4amber -i receptor.pdb -o receptor_fixed.pdb --add-missing-atoms
==================================================
Summary of pdb4amber for: receptor.pdb
===================================================
----------Chains
The following (original) chains have been found:
A
---------- Alternate Locations (Original Residues!))
The following residues had alternate locations:
None
-----------Non-standard-resnames
---------- Mising heavy atom(s)
None
1.2.3 为蛋白质去水去氢
username@computer:~/test/receptor$ pdb4amber -i receptor_fixed.pdb -o receptor_noH.pdb -y --dry
==================================================
Summary of pdb4amber for: receptor_fixed.pdb
===================================================
----------Chains
The following (original) chains have been found:
---------- Alternate Locations (Original Residues!))
The following residues had alternate locations:
None
-----------Non-standard-resnames
---------- Mising heavy atom(s)
None
1.2.4 为蛋白质重新加氢
username@computer:~/test/receptor$ reduce receptor_noH.pdb > receptor_H.pdb
reduce: version 3.3 06/02/2016, Copyright 1997-2016, J. Michael Word
Processing file: "receptor_noH.pdb"
Database of HETATM connections: "/home/program/amber20//dat/reduce_wwPDB_het_dict.txt"
VDW dot density = 16/A^2
Orientation penalty scale = 1 (100%)
Eliminate contacts within 3 bonds.
Ignore atoms with |occupancy| <= 0.01 during adjustments.
Waters ignored if B-Factor >= 40 or |occupancy| < 0.66
Aromatic rings in amino acids accept hydrogen bonds.
Building or keeping OH & SH Hydrogens.
Rotating NH3 Hydrogens.
Not processing Met methyls.
WARNING: atom H10A from HIE will be treated as hydrogen
WARNING: atom H12A from HIE will be treated as hydrogen
WARNING: atom H13A from HIE will be treated as hydrogen
WARNING: atom H17A from HIE will be treated as hydrogen
WARNING: atom H19A from HIE will be treated as hydrogen
WARNING: atom H21A from HIE will be treated as hydrogen
WARNING: atom H21B from HIE will be treated as hydrogen
WARNING: atom H22A from HIE will be treated as hydrogen
WARNING: atom H22B from HIE will be treated as hydrogen
WARNING: atom HN1A from HIE will be treated as hydrogen
WARNING: atom H10A from HIE will be treated as hydrogen
WARNING: atom H10A from HIE will be treated as hydrogen
WARNING: atom H12A from HIE will be treated as hydrogen
WARNING: atom H12A from HIE will be treated as hydrogen
WARNING: atom H13A from HIE will be treated as hydrogen
WARNING: atom H13A from HIE will be treated as hydrogen
WARNING: atom H17A from HIE will be treated as hydrogen
WARNING: atom H17A from HIE will be treated as hydrogen
WARNING: atom H19A from HIE will be treated as hydrogen
WARNING: atom H19A from HIE will be treated as hydrogen
WARNING: atom H21A from HIE will be treated as hydrogen
WARNING: atom H21A from HIE will be treated as hydrogen
WARNING: atom H21B from HIE will be treated as hydrogen
WARNING: atom H21B from HIE will be treated as hydrogen
WARNING: atom H21A from HIE will be treated as hydrogen
WARNING: atom H21B from HIE will be treated as hydrogen
WARNING: atom H22A from HIE will be treated as hydrogen
WARNING: atom H22A from HIE will be treated as hydrogen
WARNING: atom H22B from HIE will be treated as hydrogen
WARNING: atom H22B from HIE will be treated as hydrogen
WARNING: atom H22A from HIE will be treated as hydrogen
WARNING: atom H22B from HIE will be treated as hydrogen
WARNING: atom HN1A from HIE will be treated as hydrogen
WARNING: atom HN1A from HIE will be treated as hydrogen
WARNING: atom HN1A from HIE will be treated as hydrogen
WARNING: atom H22B from HIE will be treated as hydrogen
WARNING: atom H22B from HIE will be treated as hydrogen
WARNING: atom H22A from HIE will be treated as hydrogen
WARNING: atom H22A from HIE will be treated as hydrogen
WARNING: atom H22B from HIE will be treated as hydrogen
WARNING: atom H22A from HIE will be treated as hydrogen
WARNING: atom H21B from HIE will be treated as hydrogen
WARNING: atom H21B from HIE will be treated as hydrogen
WARNING: atom H21A from HIE will be treated as hydrogen
WARNING: atom H21A from HIE will be treated as hydrogen
WARNING: atom H21B from HIE will be treated as hydrogen
WARNING: atom H21A from HIE will be treated as hydrogen
WARNING: atom H19A from HIE will be treated as hydrogen
WARNING: atom H19A from HIE will be treated as hydrogen
WARNING: atom H17A from HIE will be treated as hydrogen
WARNING: atom H17A from HIE will be treated as hydrogen
WARNING: atom H13A from HIE will be treated as hydrogen
WARNING: atom H13A from HIE will be treated as hydrogen
WARNING: atom H12A from HIE will be treated as hydrogen
WARNING: atom H12A from HIE will be treated as hydrogen
WARNING: atom H10A from HIE will be treated as hydrogen
WARNING: atom H10A from HIE will be treated as hydrogen
WARNING: atom H22B from HIE will be treated as hydrogen
WARNING: atom H22A from HIE will be treated as hydrogen
WARNING: atom H21B from HIE will be treated as hydrogen
WARNING: atom H21A from HIE will be treated as hydrogen
WARNING: atom H19A from HIE will be treated as hydrogen
WARNING: atom H17A from HIE will be treated as hydrogen
WARNING: atom H13A from HIE will be treated as hydrogen
WARNING: atom H12A from HIE will be treated as hydrogen
WARNING: atom H10A from HIE will be treated as hydrogen
WARNING: atom H10A from HIE will be treated as hydrogen
WARNING: atom H12A from HIE will be treated as hydrogen
WARNING: atom H13A from HIE will be treated as hydrogen
WARNING: atom H17A from HIE will be treated as hydrogen
WARNING: atom H19A from HIE will be treated as hydrogen
WARNING: atom H21A from HIE will be treated as hydrogen
WARNING: atom H21B from HIE will be treated as hydrogen
WARNING: atom H22A from HIE will be treated as hydrogen
WARNING: atom H22B from HIE will be treated as hydrogen
WARNING: atom HN1A from HIE will be treated as hydrogen
WARNING: atom H10A from HIE will be treated as hydrogen
WARNING: atom H10A from HIE will be treated as hydrogen
WARNING: atom H12A from HIE will be treated as hydrogen
WARNING: atom H12A from HIE will be treated as hydrogen
WARNING: atom H13A from HIE will be treated as hydrogen
WARNING: atom H13A from HIE will be treated as hydrogen
WARNING: atom H17A from HIE will be treated as hydrogen
WARNING: atom H17A from HIE will be treated as hydrogen
WARNING: atom H19A from HIE will be treated as hydrogen
WARNING: atom H19A from HIE will be treated as hydrogen
WARNING: atom H21A from HIE will be treated as hydrogen
WARNING: atom H21A from HIE will be treated as hydrogen
WARNING: atom H21B from HIE will be treated as hydrogen
WARNING: atom H21B from HIE will be treated as hydrogen
WARNING: atom H21A from HIE will be treated as hydrogen
WARNING: atom H21B from HIE will be treated as hydrogen
WARNING: atom H22A from HIE will be treated as hydrogen
WARNING: atom H22A from HIE will be treated as hydrogen
WARNING: atom H22B from HIE will be treated as hydrogen
WARNING: atom H22B from HIE will be treated as hydrogen
WARNING: atom H22A from HIE will be treated as hydrogen
WARNING: atom H22B from HIE will be treated as hydrogen
WARNING: atom HN1A from HIE will be treated as hydrogen
WARNING: atom HN1A from HIE will be treated as hydrogen
WARNING: atom HN1A from HIE will be treated as hydrogen
WARNING: atom H22B from HIE will be treated as hydrogen
WARNING: atom H22B from HIE will be treated as hydrogen
WARNING: atom H22A from HIE will be treated as hydrogen
WARNING: atom H22A from HIE will be treated as hydrogen
WARNING: atom H22B from HIE will be treated as hydrogen
WARNING: atom H22A from HIE will be treated as hydrogen
WARNING: atom H21B from HIE will be treated as hydrogen
WARNING: atom H21B from HIE will be treated as hydrogen
WARNING: atom H21A from HIE will be treated as hydrogen
WARNING: atom H21A from HIE will be treated as hydrogen
WARNING: atom H21B from HIE will be treated as hydrogen
WARNING: atom H21A from HIE will be treated as hydrogen
WARNING: atom H19A from HIE will be treated as hydrogen
WARNING: atom H19A from HIE will be treated as hydrogen
WARNING: atom H17A from HIE will be treated as hydrogen
WARNING: atom H17A from HIE will be treated as hydrogen
WARNING: atom H13A from HIE will be treated as hydrogen
WARNING: atom H13A from HIE will be treated as hydrogen
WARNING: atom H12A from HIE will be treated as hydrogen
WARNING: atom H12A from HIE will be treated as hydrogen
WARNING: atom H10A from HIE will be treated as hydrogen
WARNING: atom H10A from HIE will be treated as hydrogen
WARNING: atom H22B from HIE will be treated as hydrogen
WARNING: atom H22A from HIE will be treated as hydrogen
WARNING: atom H21B from HIE will be treated as hydrogen
WARNING: atom H21A from HIE will be treated as hydrogen
WARNING: atom H19A from HIE will be treated as hydrogen
WARNING: atom H17A from HIE will be treated as hydrogen
WARNING: atom H13A from HIE will be treated as hydrogen
WARNING: atom H12A from HIE will be treated as hydrogen
WARNING: atom H10A from HIE will be treated as hydrogen
Singles(size 22): 1 ALA N : 15 THR OG1 : 16 THR OG1 : 21 SER OG
: 33 LYS NZ : 34 TYR OH : 36 LYS NZ : 38 LYS NZ : 45 THR OG1
: 48 SER OG : 49 TYR OH : 50 TYR OH : 55 TYR OH : 62 SER OG
: 63 THR OG1 : 66 SER OG : 68 THR OG1 : 71 LYS NZ : 72 LYS NZ
: 75 TYR OH : 77 THR OG1 : 81 THR OG1
orientation 3: 1 ALA N :NH3+ 149: bump=0.000, HB=0.056, total=0.056
orientation 1: 15 THR OG1 : rot 180: bump=0.000, HB=0.026, total=0.026
orientation 1: 16 THR OG1 : rot 180: bump=0.000, HB=0.000, total=0.000
orientation 3: 21 SER OG : rot -83: bump=-0.016, HB=1.355, total=1.339
orientation 1: 33 LYS NZ :NH3+ 180: bump=0.000, HB=0.000, total=0.000
orientation 1: 34 TYR OH : rot 180: bump=-1.278, HB=0.000, total=-1.278
orientation 1: 36 LYS NZ :NH3+ 180: bump=0.000, HB=0.000, total=0.000
orientation 4: 38 LYS NZ :NH3+ -111: bump=0.000, HB=0.172, total=0.172
orientation 2: 45 THR OG1 : rot 71: bump=0.000, HB=0.448, total=0.448
orientation 2: 48 SER OG : rot 77: bump=0.000, HB=0.012, total=0.012
orientation 11: 49 TYR OH : rot -66: bump=-0.590, HB=0.695, total=0.105
orientation 8: 50 TYR OH : rot -15: bump=0.000, HB=1.195, total=1.195
orientation 1: 55 TYR OH : rot 180: bump=-0.194, HB=0.000, total=-0.194
orientation 1: 62 SER OG : rot 131: bump=0.000, HB=0.317, total=0.317
orientation 1: 63 THR OG1 : rot 180: bump=0.000, HB=0.000, total=0.000
orientation 1: 66 SER OG : rot 180: bump=-0.054, HB=0.000, total=-0.054
orientation 1: 68 THR OG1 : rot 138: bump=-0.113, HB=2.146, total=2.033
orientation 1: 71 LYS NZ :NH3+ 180: bump=0.000, HB=0.000, total=0.000
orientation 1: 72 LYS NZ :NH3+ 180: bump=0.000, HB=0.000, total=0.000
orientation 1: 75 TYR OH : rot 180: bump=0.000, HB=0.000, total=0.000
orientation 1: 77 THR OG1 : rot 180: bump=0.000, HB=0.000, total=0.000
orientation 3: 81 THR OG1 : rot 63: bump=0.000, HB=0.786, total=0.786
Found 0 hydrogens (0 hets)
Standardized 0 hydrogens (0 hets)
Added 652 hydrogens (0 hets)
Removed 0 hydrogens (0 hets)
Adjusted 18 group(s)
If you publish work which uses reduce, please cite:
Word, et. al. (1999) J. Mol. Biol. 285, 1735-1747.
For more information see http://kinemage.biochem.duke.edu
1.2.5 为蛋白质重新编号
username@computer:~/test/receptor$ pdb4amber -i receptor_H.pdb -o receptor_H_renum.pdb
==================================================
Summary of pdb4amber for: receptor_H.pdb
===================================================
----------Chains
The following (original) chains have been found:
---------- Alternate Locations (Original Residues!))
The following residues had alternate locations:
None
-----------Non-standard-resnames
---------- Mising heavy atom(s)
None
二、生成复合物的拓扑文件和坐标文件
username@computer:~/test$ vi tleap.in
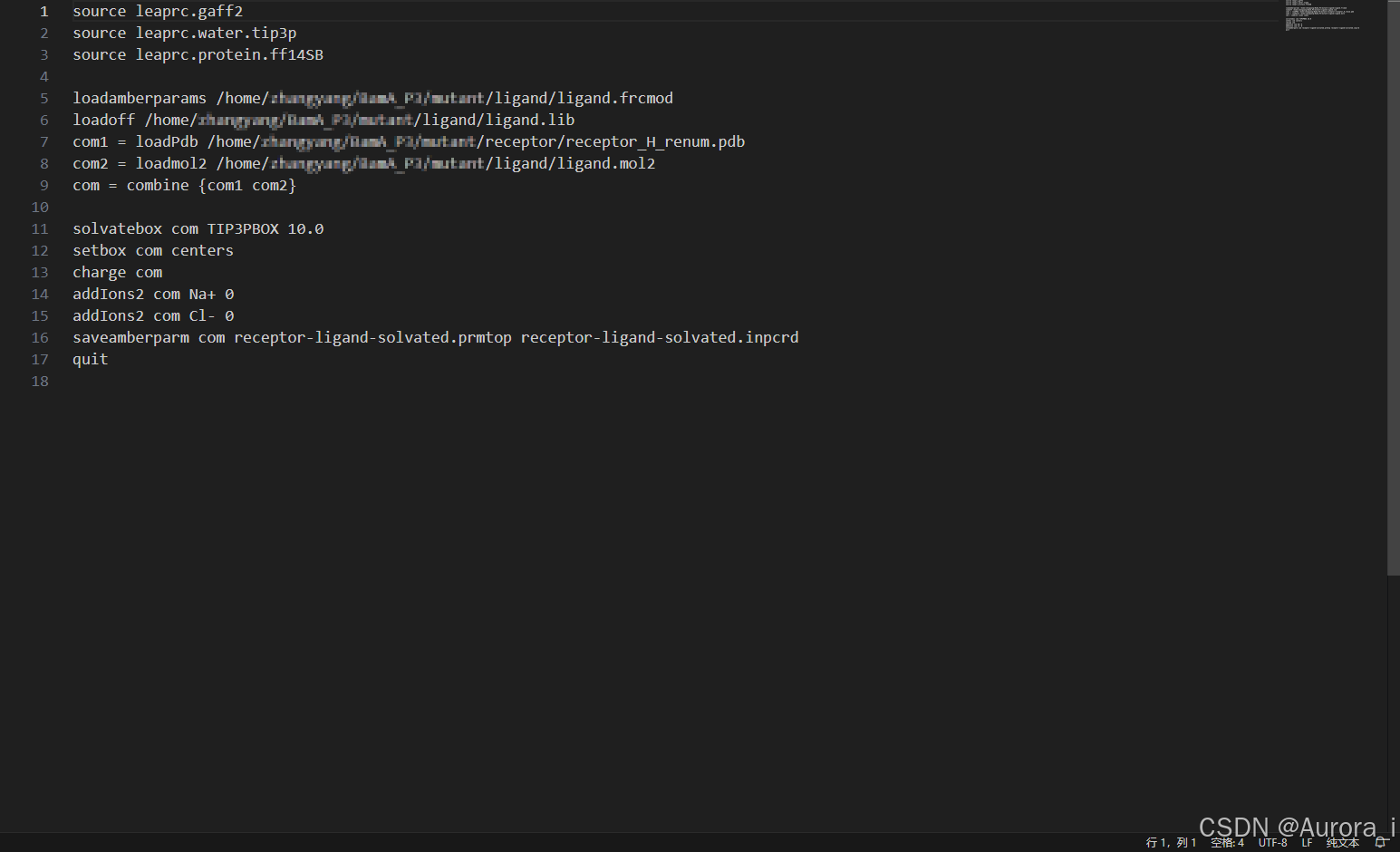
username@computer:~/test$ tleap -f tleap.in
-I: Adding /home/program/amber20/dat/leap/prep to search path.
-I: Adding /home/program/amber20/dat/leap/lib to search path.
-I: Adding /home/program/amber20/dat/leap/parm to search path.
-I: Adding /home/program/amber20/dat/leap/cmd to search path.
-f: Source tleap.in.
Welcome to LEaP!
(no leaprc in search path)
Sourcing: ./tleap.in
----- Source: /home/program/amber20/dat/leap/cmd/leaprc.gaff2
----- Source of /home/program/amber20/dat/leap/cmd/leaprc.gaff2 done
Log file: ./leap.log
Loading parameters: /home/program/amber20/dat/leap/parm/gaff2.dat
Reading title:
AMBER General Force Field for organic molecules (Version 2.11, May 2016)
----- Source: /home/program/amber20/dat/leap/cmd/leaprc.water.tip3p
----- Source of /home/program/amber20/dat/leap/cmd/leaprc.water.tip3p done
Loading library: /home/program/amber20/dat/leap/lib/atomic_ions.lib
Loading library: /home/program/amber20/dat/leap/lib/solvents.lib
Loading parameters: /home/program/amber20/dat/leap/parm/frcmod.tip3p
Reading force field modification type file (frcmod)
Reading title:
This is the additional/replacement parameter set for TIP3P water
Loading parameters: /home/program/amber20/dat/leap/parm/frcmod.ions1lm_126_tip3p
Reading force field modification type file (frcmod)
Reading title:
Li/Merz ion parameters of monovalent ions for TIP3P water model (12-6 normal usage set)
Loading parameters: /home/program/amber20/dat/leap/parm/frcmod.ionsjc_tip3p
Reading force field modification type file (frcmod)
Reading title:
Monovalent ion parameters for Ewald and TIP3P water from Joung & Cheatham JPCB (2008)
Loading parameters: /home/program/amber20/dat/leap/parm/frcmod.ions234lm_126_tip3p
Reading force field modification type file (frcmod)
Reading title:
Li/Merz ion parameters of divalent to tetravalent ions for TIP3P water model (12-6 normal usage set)
----- Source: /home/program/amber20/dat/leap/cmd/leaprc.protein.ff14SB
----- Source of /home/program/amber20/dat/leap/cmd/leaprc.protein.ff14SB done
Log file: ./leap.log
Loading parameters: /home/program/amber20/dat/leap/parm/parm10.dat
Reading title:
PARM99 + frcmod.ff99SB + frcmod.parmbsc0 + OL3 for RNA
Loading parameters: /home/program/amber20/dat/leap/parm/frcmod.ff14SB
Reading force field modification type file (frcmod)
Reading title:
ff14SB protein backbone and sidechain parameters
Loading library: /home/program/amber20/dat/leap/lib/amino12.lib
Loading library: /home/program/amber20/dat/leap/lib/aminoct12.lib
Loading library: /home/program/amber20/dat/leap/lib/aminont12.lib
Loading parameters: /home/username/test/ligand/ligand.frcmod
Reading force field modification type file (frcmod)
Reading title:
Remark line goes here
Loading library: /home/username/test/ligand/ligand.lib
Loading PDB file: /home/username/test/receptor/receptor_H_renum.pdb
total atoms in file: 1427
Leap added 14 missing atoms according to residue templates:
14 H / lone pairs
Loading Mol2 file: /home/username/test/ligand/ligand.mol2
Reading MOLECULE named UNL
Solute vdw bounding box: 30.498 47.289 39.472
Total bounding box for atom centers: 50.498 67.289 59.472
Solvent unit box: 18.774 18.774 18.774
Total vdw box size: 53.760 70.410 62.528 angstroms.
Volume: 236686.993 A^3
Total mass 109663.946 amu, Density 0.769 g/cc
Added 5497 residues.
removing previous box..
Box dimensions: 50.270201 67.267982 59.431706
Total unperturbed charge: -1.998000
Total perturbed charge: -1.998000
2 Na+ ions required to neutralize.
Adding 2 counter ions to "com" using 1A grid
Grid extends from solute vdw + 1.37 to 7.28
Resolution: 1.00 Angstrom.
Calculating grid charges
Placed Na+ in com at (6.65, 21.40, 5.92).
Placed Na+ in com at (1.65, -3.60, 11.92).
Done adding ions.
0.002000 0 1 0
0 Cl- ion required to neutralize.
Adding 0 counter ions to "com" using 1A grid
Checking Unit.
Building topology.
Building atom parameters.
Building bond parameters.
Building angle parameters.
Building proper torsion parameters.
Building improper torsion parameters.
total 318 improper torsions applied
Building H-Bond parameters.
Incorporating Non-Bonded adjustments.
Not Marking per-residue atom chain types.
Marking per-residue atom chain types.
(Residues lacking connect0/connect1 -
these don't have chain types marked:
res total affected
CGLY 1
NALA 1
UNL 1
WAT 5497
)
(no restraints)
Quit
Exiting LEaP: Errors = 0; Warnings = 0; Notes = 0.
根据生成的拓扑文件和坐标文件重新生成复合物 pdb 文件,检查是否有误
username@computer:~/test$ ambpdb -p receptor-ligand-solvated.prmtop -c receptor-ligand-solvated.inpcrd > complex.pdb
三、Amber 分子动力学模拟
username@computer:~/test$ mv receptor-ligand-solvated.inpcrd /home/username/test/MD
username@computer:~/test$ mv receptor-ligand-solvated.prmtop /home/username/test/MD
username@computer:~/test$ cd MD
username@computer:~/test/MD$ ls
do_HMR.in eq2.in eq4.in min1.in min3.in output.sh receptor-ligand-solvated.inpcrd
eq1.in eq3.in heat.in min2.in min4.in prod1.in receptor-ligand-solvated.prmtop
以下是对各个 in 文件及 sh 文件的展开:
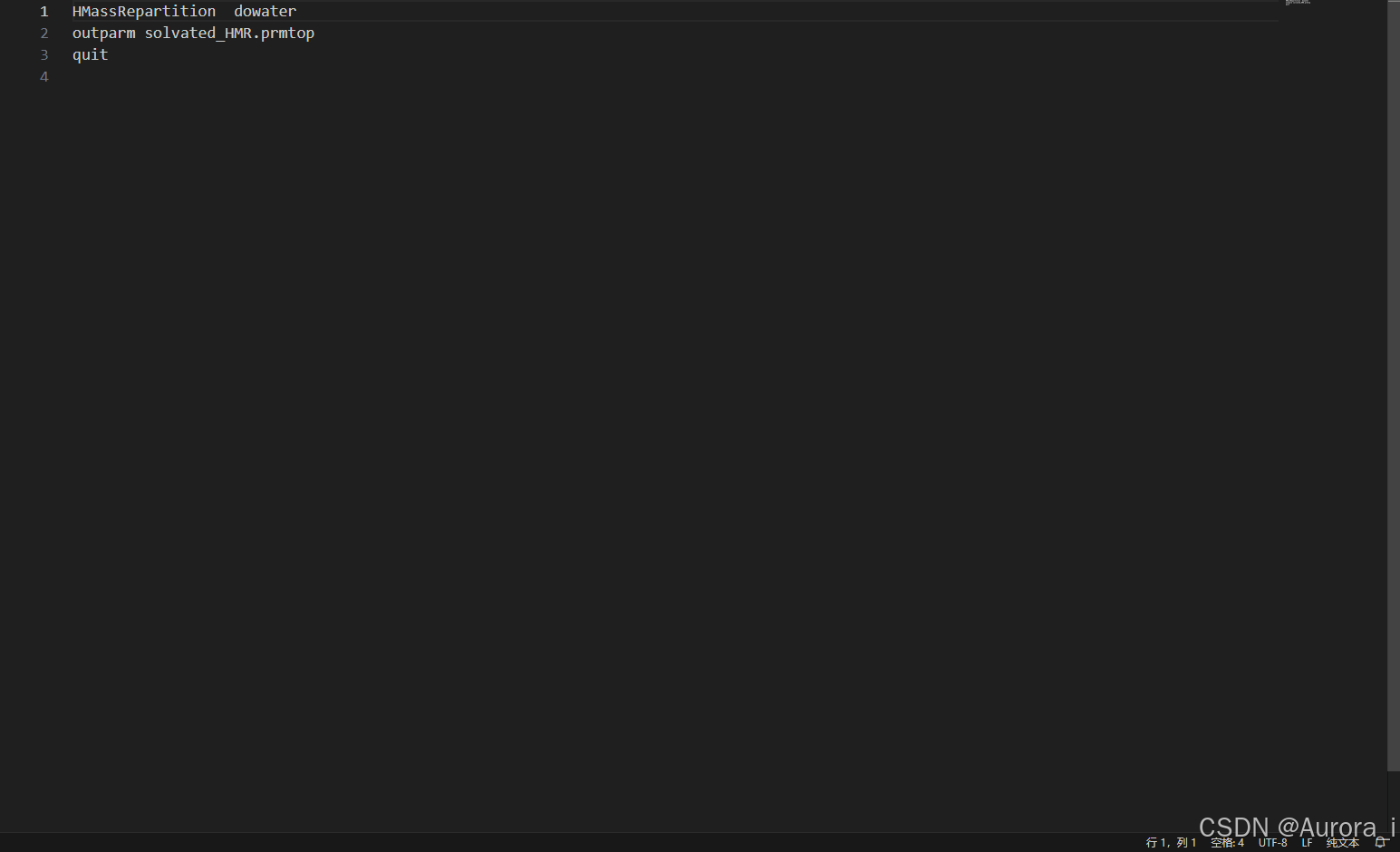
do_HMR.in
eq1.in
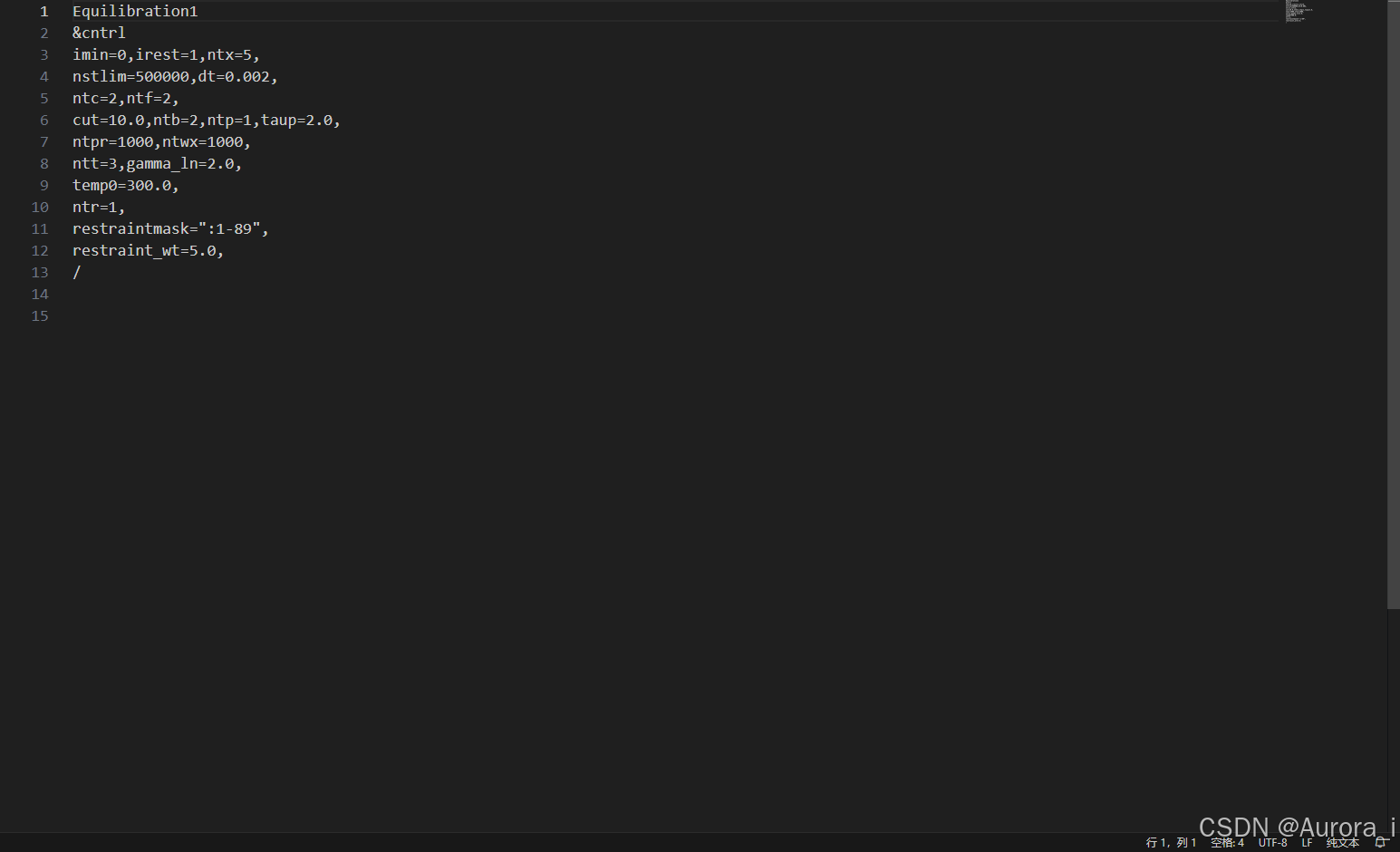
注意:restraintmask 这一参数项请选择上述 complex.pdb 文件中蛋白质的残基编号
eq2.in
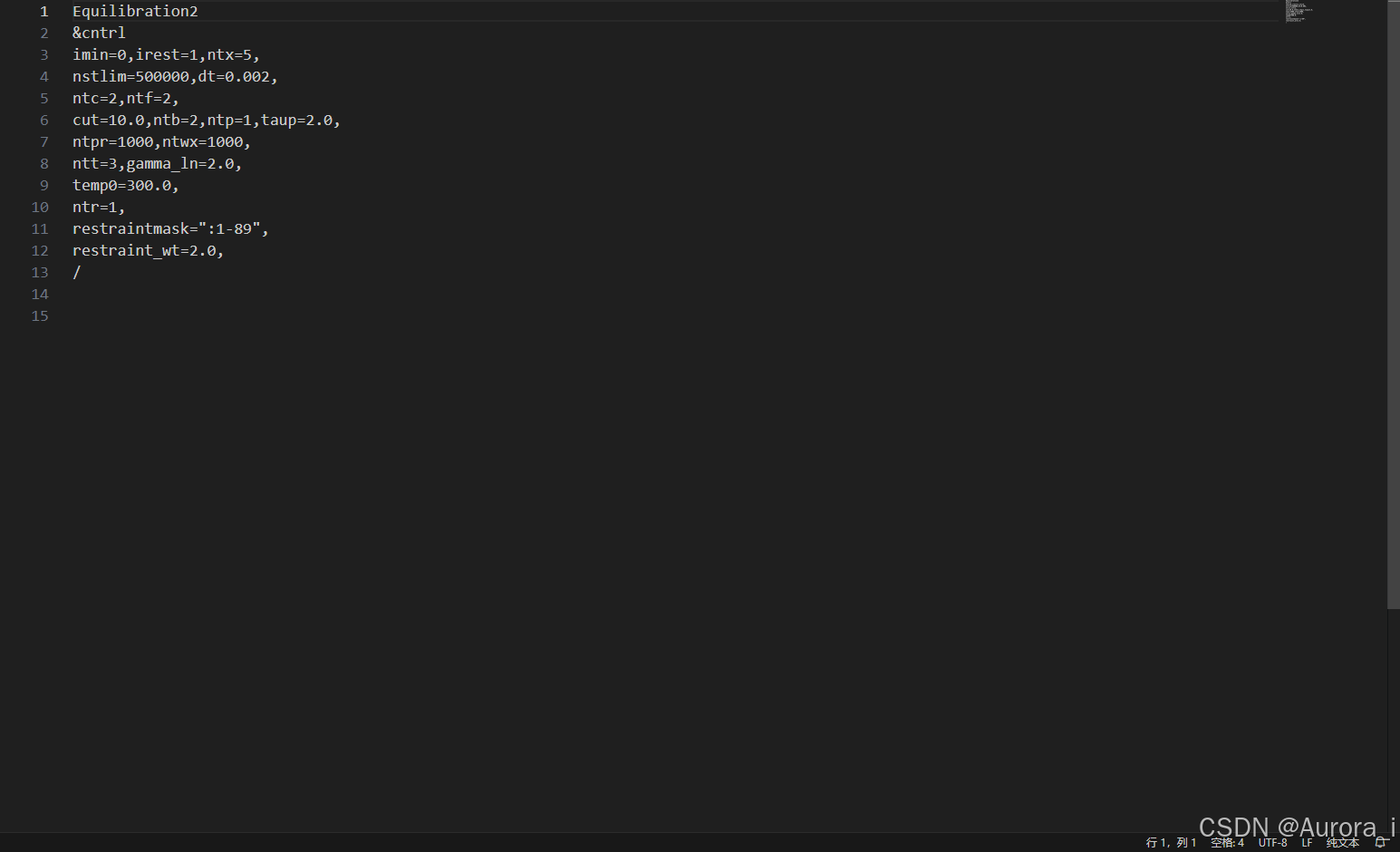
eq3.in
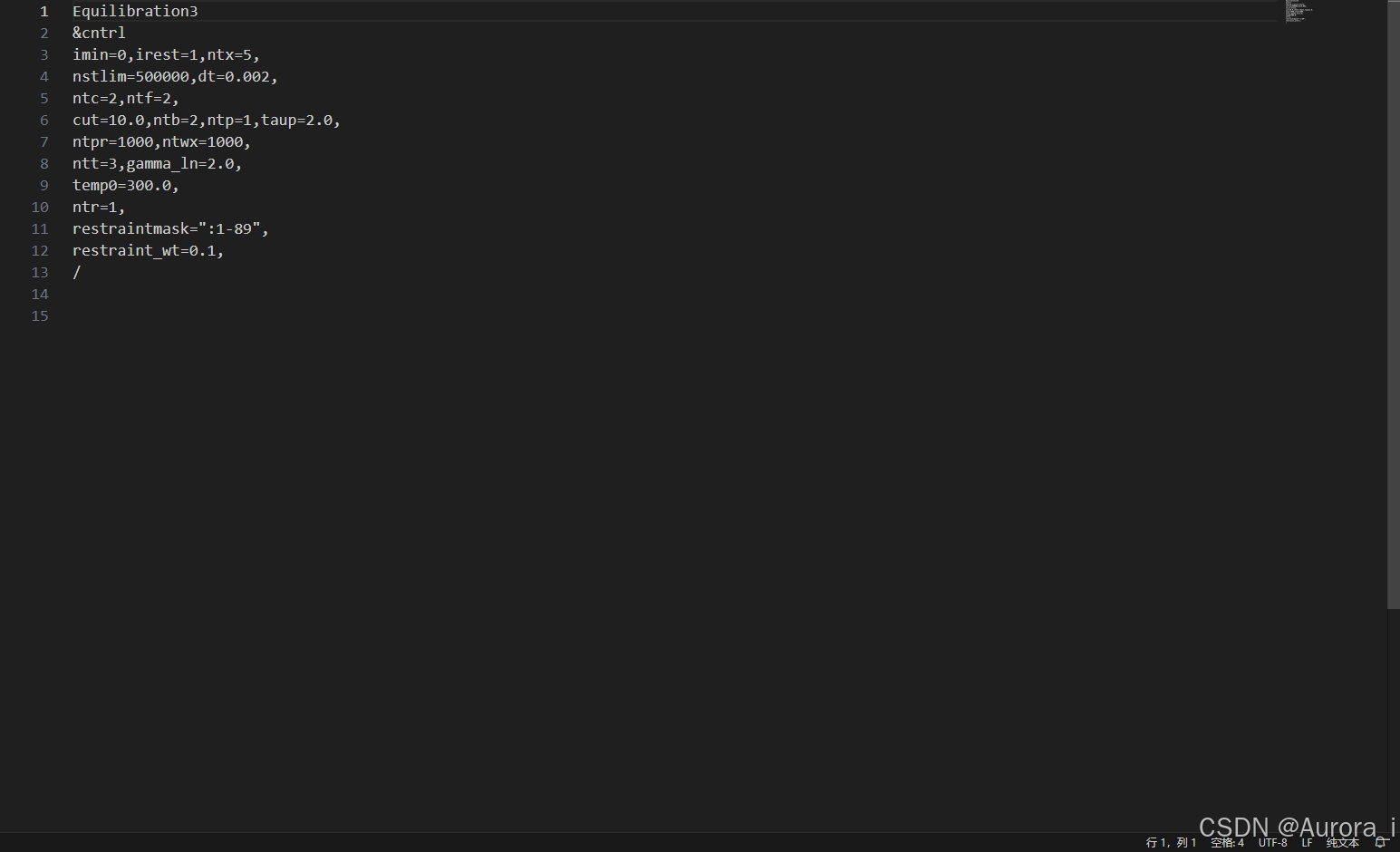
eq4.in
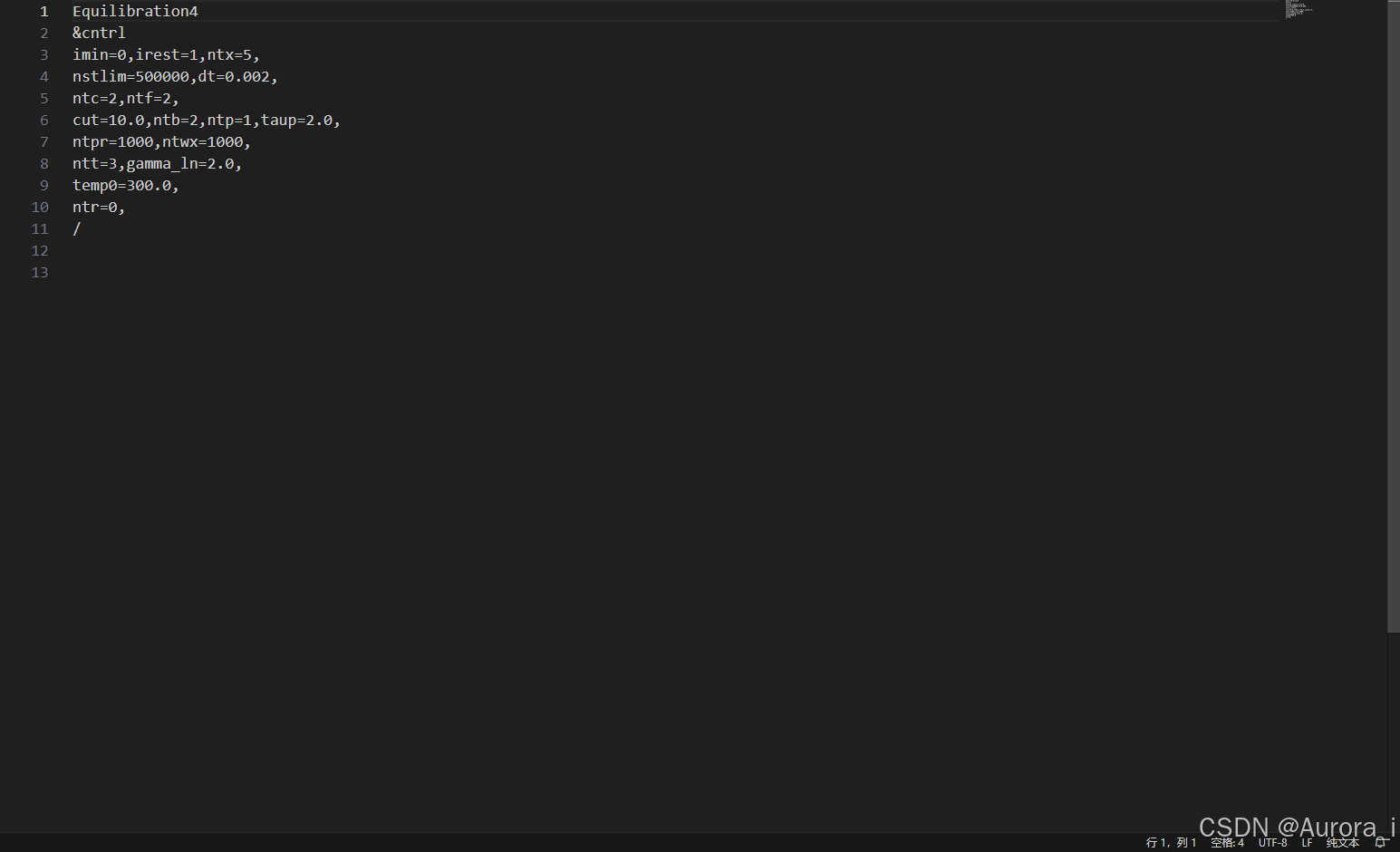
heat.in
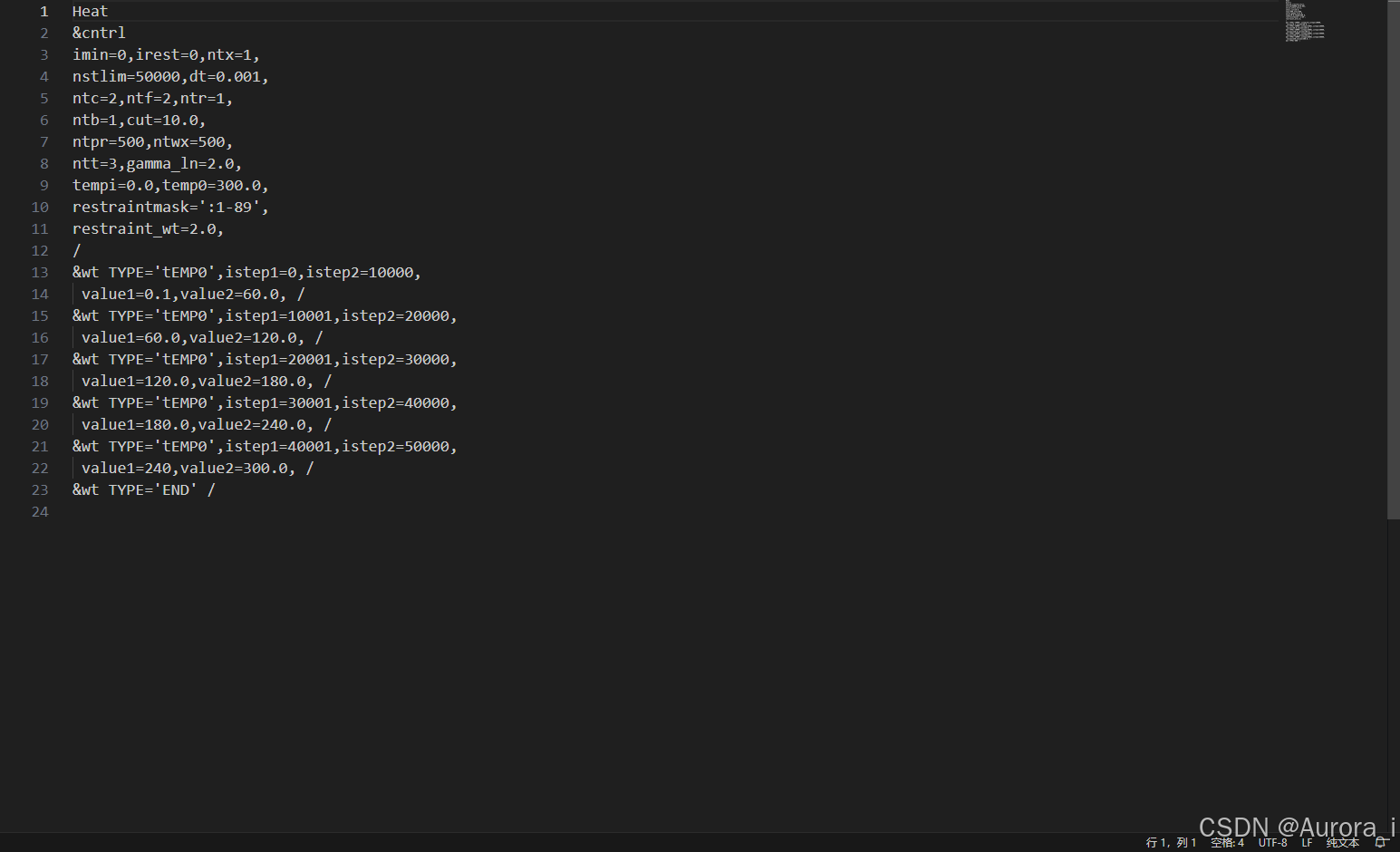
min1.in
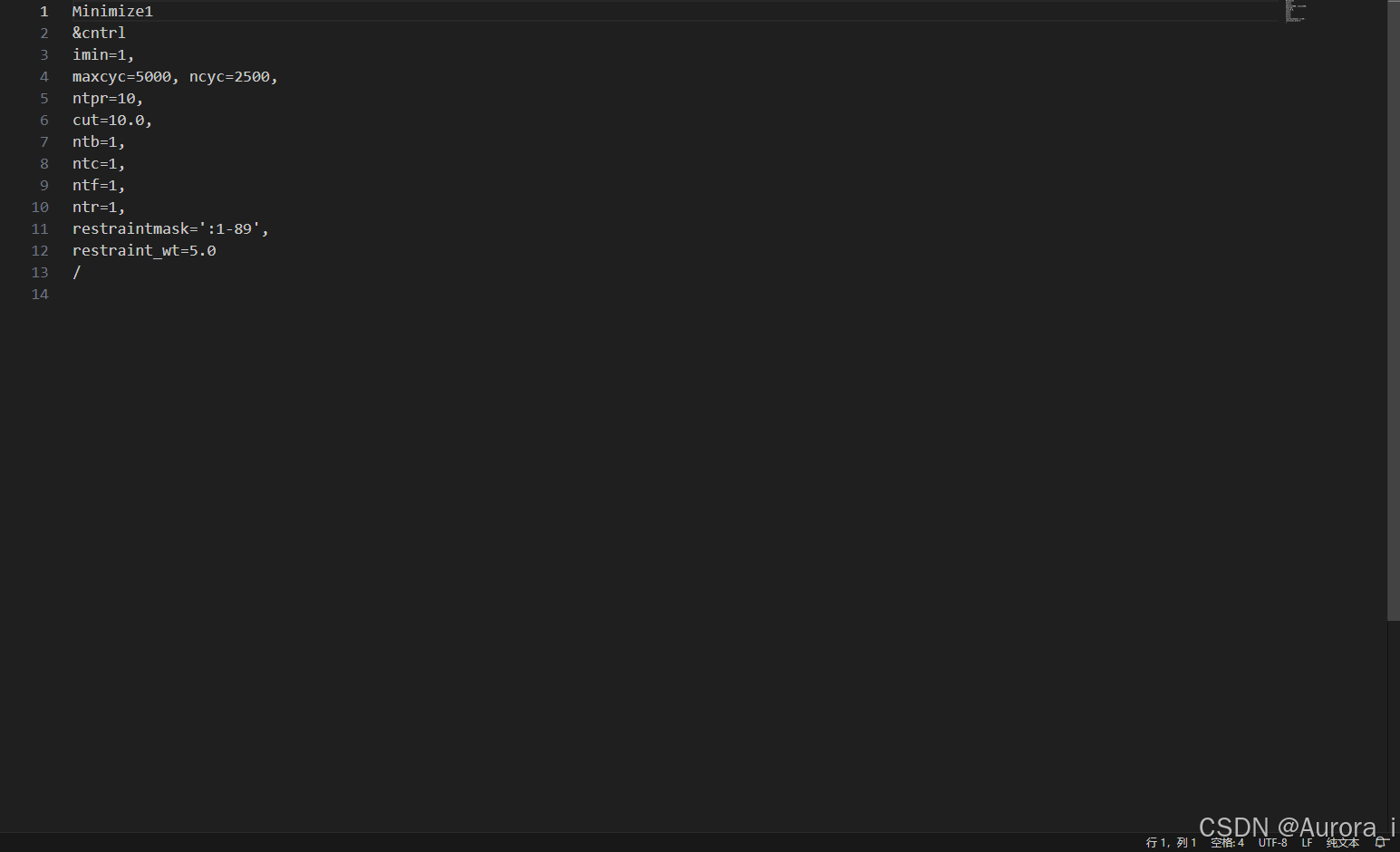
min2.in
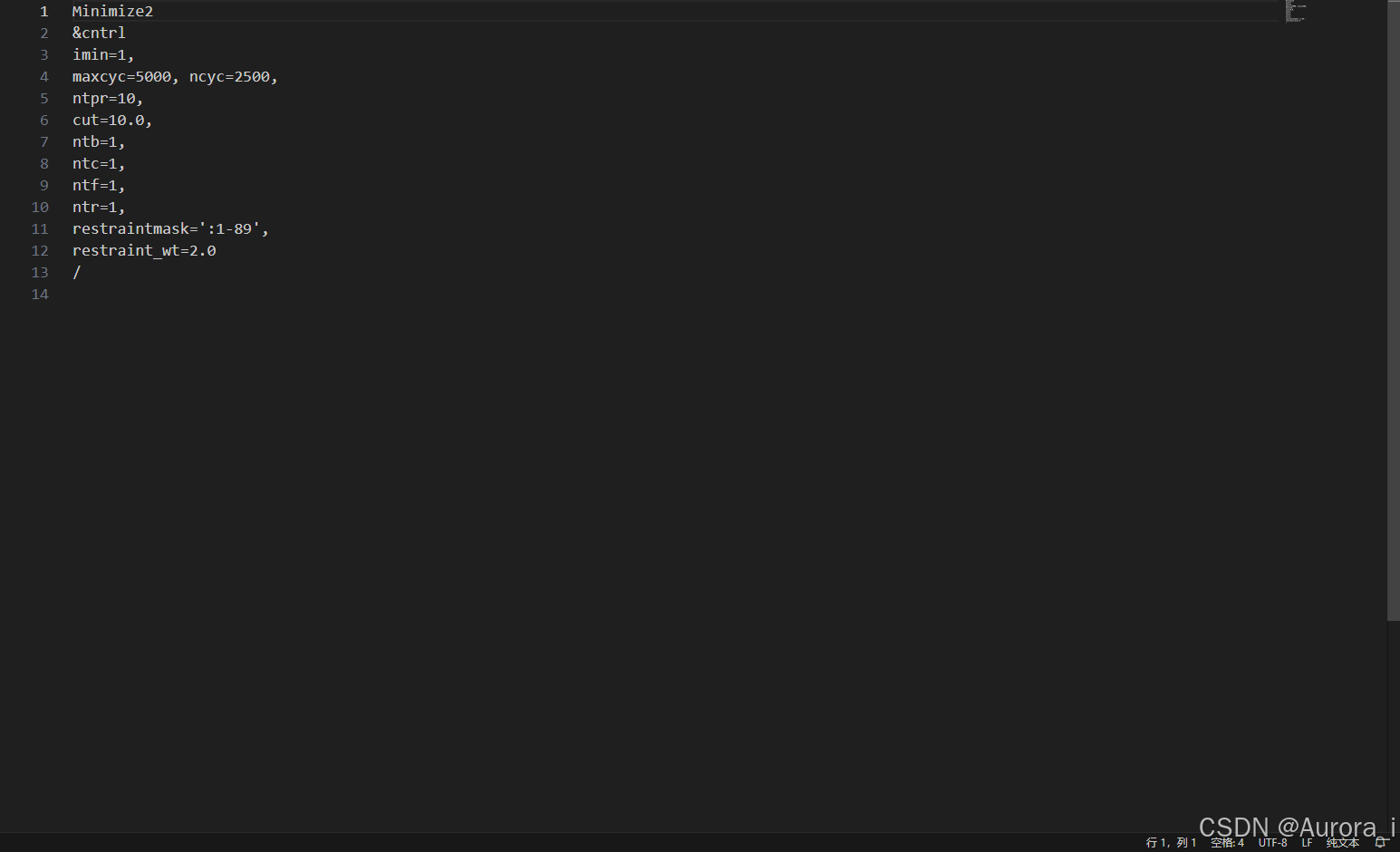
min3.in
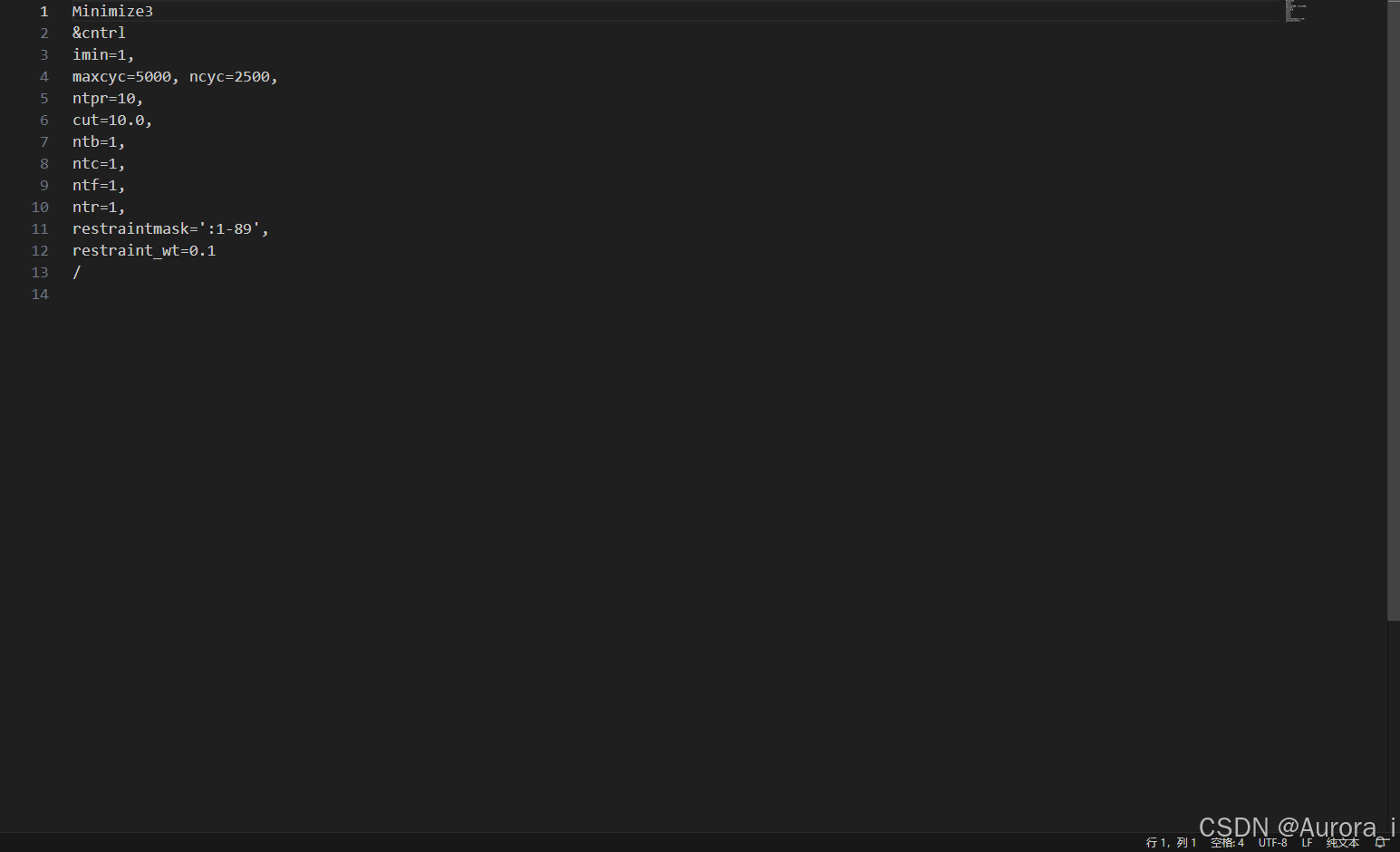
min4.in

prod1.in
注意:nstlim 参数项代表步数,dt 参数项代表时间步长(ps),那么这里表示总共进行 25000000 * 0.002 = 50000 ps = 50 ns
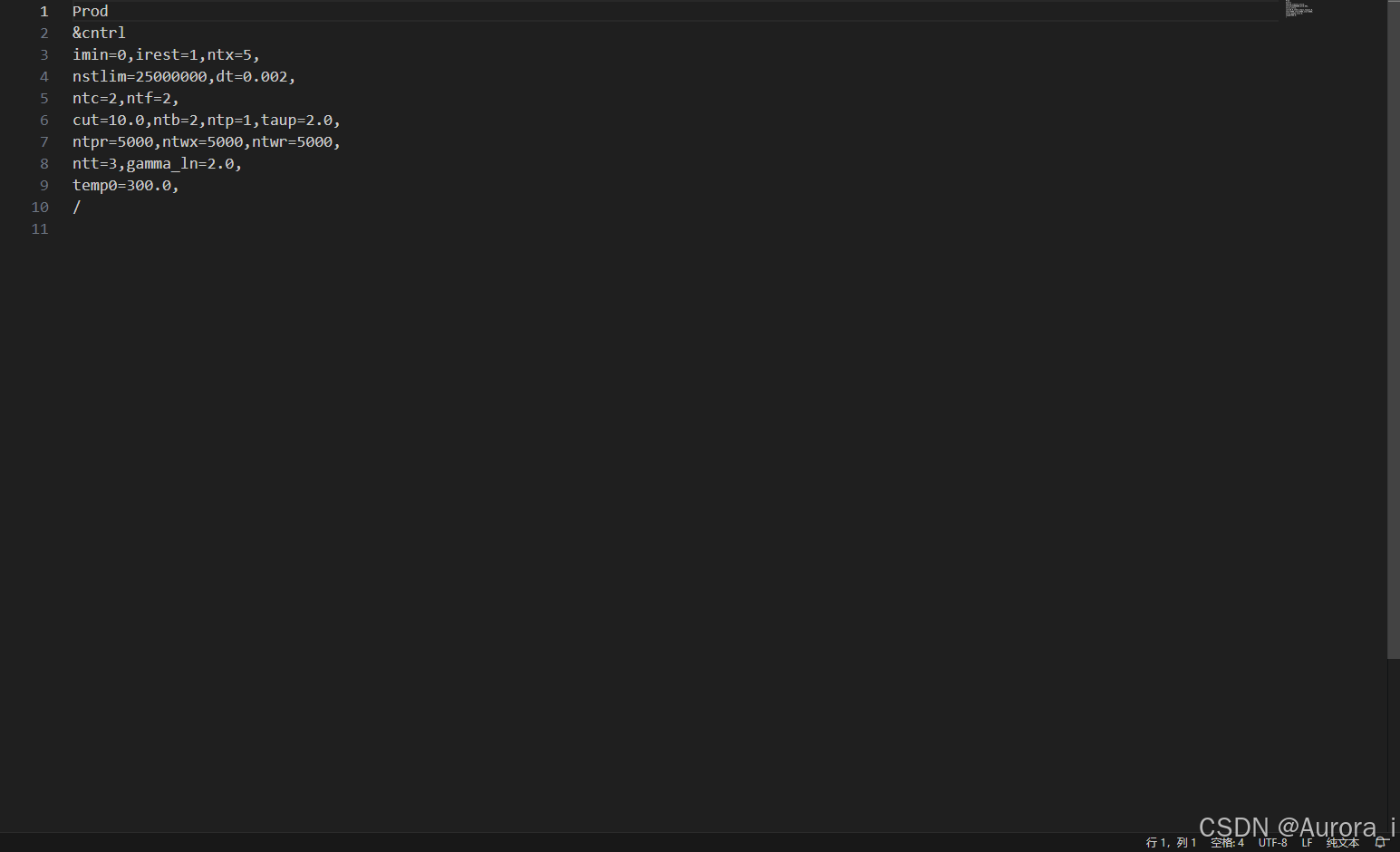
output.sh
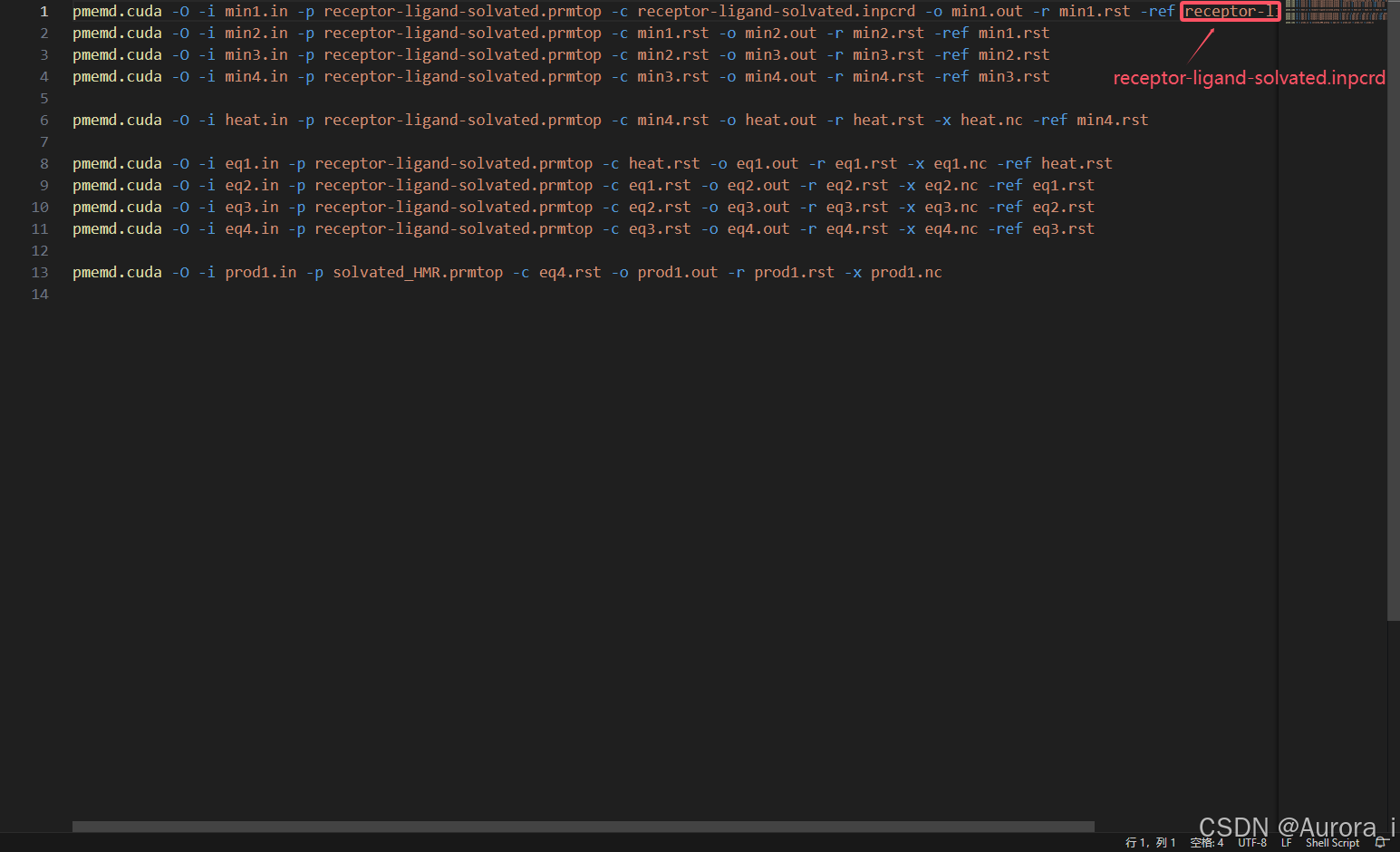
复合物氢质量的重新分配:
username@computer:~/test/MD$ parmed -p receptor-ligand-solvated.prmtop -i do_HMR.in
进行分子动力学模拟:
username@computer:~/test/MD$ nohup sh output.sh &
程序运行完成后:
username@computer:~/test/MD$ ls
do_HMR.in eq1.rst eq2.rst eq3.rst eq4.rst heat.rst min1.rst min3.in min4.out prod1.in receptor-ligand-solvated.inpcrd
eq1.in eq2.in eq3.in eq4.in heat.in mdinfo min2.in min3.out min4.rst prod1.nc receptor-ligand-solvated.prmtop
eq1.nc eq2.nc eq3.nc eq4.nc heat.nc min1.in min2.out min3.rst nohup.out prod1.out solvated_HMR.prmtop
eq1.out eq2.out eq3.out eq4.out heat.out min1.out min2.rst min4.in output.sh prod1.rst
至此,Amber20 蛋白质-小分子复合物的分子动力学模拟已全部完成!


















 779
779

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









