






BLAST(Basic Local Alignment Search Tool,基本局部比对搜索工具)是一种广泛用于生物信息学的序列比对算法,主要用于核酸或蛋白质序列在数据库中的搜索与比对。其核心原理是基于局部比对的启发式算法,能够快速识别高相似度的序列区域,而无需进行全局比对。

以下是BLAST的介绍:
1. BLAST的基本流程
BLAST的比对过程主要分为五个步骤:
- 种子匹配(Seed Matching)
- 种子扩展(Extension)
- HSP筛选(High-scoring Segment Pairs, HSPs)
- 统计评估(Scoring & Evaluation)
- 结果输出(Output & Reporting)
2. 详细比对原理
(1)种子匹配(Seed Matching)
BLAST使用k-mer(短字词)索引法快速寻找目标数据库中可能的匹配区域:
-
先将查询序列切割成多个k-mer(k-字母短词),默认k值:
- 蛋白序列
:默认 k = 3(称为三肽)
- DNA序列
:默认 k = 11
- 蛋白序列
-
然后,将这些短词在数据库序列中寻找完全匹配或近似匹配的区域。
-
通过**索引表(Hash Table)**快速查找候选区域,避免直接进行全局比对。
👉 例子:
-
查询序列(蛋白):
MKTLLL - k = 3,则生成短词:
MKT, KTL, TLL, LLL
-
在数据库中查找是否存在这些短词的匹配项。
(2)种子扩展(Extension)
当找到匹配的**种子序列(seed word)**后,BLAST会尝试向两侧延伸,以获得更长的匹配区域:
-
采用动态规划方法(类似Smith-Waterman算法),在匹配点两侧扩展比对。
-
计算比对分数(alignment score):
- 匹配(Match)
:+得分
- 错配(Mismatch)
:-罚分
- 空位(Gap)
:-罚分
- 匹配(Match)
-
直到比对分数下降到一定阈值(低于“下降值” drop threshold),停止扩展。
👉 例子:
Query: ...MKTLLLSIV...
Database: ...MKTLLLSLV...
比对过程中:
MKTLLLSIV
MKTLLLSLV (仅S -> I 替换)
扩展过程在检测到替换(S -> I)后继续,但如果进一步错配过多,扩展会停止。
(3)HSP筛选(High-scoring Segment Pairs, HSPs)
-
**HSP(高得分片段对)**是指具有较高匹配得分的局部比对区域。
-
BLAST使用**阈值(threshold)**来过滤低质量比对:
-
仅保留高得分的HSP。
-
HSP通常是比对结果的核心,BLAST会基于这些HSP进行进一步统计分析。
-
(4)统计评估(Scoring & Evaluation)
BLAST采用统计学方法评估比对的可靠性,主要计算:
1. Bit Score(比特分数)
比特分数用于比较不同BLAST运行之间的可比性:
-
计算公式:
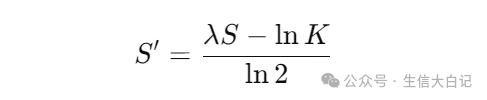
-
其中:
- S:原始比对得分
- λ(Lambda)和 K:基于数据库和查询序列计算的参数
-
Bit Score越高,代表比对越可靠。
2. E-value(期望值)
E-value 表示随机匹配出现在数据库中的概率,计算公式:
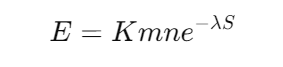
其中:
- m:查询序列长度
- n:数据库中序列总长度
- λ(Lambda)和 K:校正参数
- S:比对得分
解释:
- E-value 越小,说明比对越可靠。
-
一般情况下,E < 1e-5(0.00001) 的比对结果被认为是显著的。
(5)结果输出(Output & Reporting)
BLAST最终输出:
- 匹配序列ID
- 比对得分(Bit Score)
- E-value
- HSP区域的比对细节
- 查询序列与数据库序列的比对图示
3. BLAST的优化
(1)不同BLAST算法
BLAST 有多个变体:
| BLAST 版本 | 适用对象 | 说明 |
|---|---|---|
| BLASTN | DNA vs DNA | 用于核酸比对,适用于长序列 |
| BLASTP | 蛋白 vs 蛋白 | 适用于蛋白质序列比对 |
| BLASTX | DNA vs 蛋白 | 查询序列为DNA,翻译成蛋白后进行比对 |
| TBLASTN | 蛋白 vs DNA | 数据库序列为DNA,翻译成蛋白后比对 |
| TBLASTX | DNA vs DNA | 双方均翻译成蛋白,再进行比对 |
(2)参数优化
用户可以通过调整word size(种子长度)、**gap penalty(缺口罚分)**等参数优化比对:
- 更小的word size
(如BLASTN设为 7)提高灵敏度,适用于短序列比对。
- 调整gap penalty
可以优化插入缺失(Indel)检测。
4. BLAST的局限性
虽然BLAST比对快速高效,但仍有局限:
- 局部比对
:不适用于全局比对,可能错过远端关系。
- 对大数据的计算要求高
:BLAST比对百万序列时计算量较大。
- 对长序列不够精确
:不适合基因组级别的长序列比对(推荐使用BWA、Bowtie等)。
5. 总结
-
BLAST基于启发式方法,先寻找短词匹配,再进行扩展比对,并通过统计方法评估显著性。
-
计算E-value 评估比对的可靠性,Bit Score 进行评分比较。
-
适用于DNA、RNA、蛋白等生物序列的比对,在基因注释、进化分析、功能预测等方面广泛应用。
BLAST比对的基本命令流程
BLAST(Basic Local Alignment Search Tool)主要用于DNA、RNA 和蛋白质序列的比对,能够快速搜索数据库并找到相似序列。以下是BLAST比对的完整命令流程,包括安装、数据库构建、查询比对、结果解析等步骤。
1. 安装BLAST
如果你尚未安装BLAST,可以使用以下方法进行安装:
(1)Linux / macOS
使用conda或apt安装:
# 使用conda安装(推荐)
conda install -c bioconda blast
# Ubuntu/Debian系统
sudo apt update && sudo apt install ncbi-blast+(2)Windows
可以从NCBI官方网站下载:https://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/
2. 准备数据库
BLAST比对需要一个数据库作为参考,通常可以选择:
- NCBI数据库
(如
nt、nr) - 自定义序列数据库
(1)下载NCBI公共数据库
如果要比对核酸序列,可以使用nt(核酸库):
如果要比对蛋白序列,可以使用nr(蛋白库):
update_blastdb.pl --decompress nr(2)自定义BLAST数据库
如果要比对自己的基因组或蛋白序列,需要先用makeblastdb创建数据库。
① 创建DNA数据库
假设你有一个参考基因组genome.fa,可以用以下命令创建BLAST数据库:
makeblastdb -in genome.fa -dbtype nucl -out genome_db
参数解析:
-in genome.fa:输入的FASTA文件
-dbtype nucl:指定数据库类型(核酸
nucl或 蛋白prot)-out genome_db:输出数据库名称
② 创建蛋白数据库
如果比对的是蛋白序列:
makeblastdb -in proteins.fasta -dbtype prot -out protein_db
3. 运行BLAST比对
BLAST提供多个程序,适用于不同类型的比对:
| BLAST工具 | 适用情况 |
|---|---|
| blastn | DNA vs DNA |
| blastp | 蛋白 vs 蛋白 |
| blastx | DNA vs 蛋白(先翻译) |
| tblastn | 蛋白 vs DNA(数据库翻译) |
| tblastx | DNA vs DNA(双向翻译) |
(1)DNA序列比对(blastn)
如果你的查询序列是DNA,需要比对到基因组,可以使用blastn:
blastn -query query.fa -db genome_db -out results.txt -evalue 1e-5 -outfmt 6 -num_threads 4参数解析:
-query query.fa:查询序列
-db genome_db:参考数据库
-evalue 1e-5:E-value阈值(值越小结果越可靠)
-outfmt 6:输出格式(6表示表格格式)
-num_threads 4:使用4个线程加速
示例输出(表格格式-outfmt 6):
query1 scaffold_1 97.8 300 0 0 100 400 5000 5300 1e-50 200query2 scaffold_2 95.6 250 3 1 50 300 8000 8250 2e-40 180
字段解析:
-
Query ID(查询序列名称)
-
Subject ID(比对的目标序列)
-
相似度(% identity)
-
比对长度(alignment length)
-
错配数(mismatches)
-
缺失数(gap openings)
-
Query起始位点
-
Query终止位点
-
Subject起始位点
-
Subject终止位点
-
E-value
-
Bit score
(2)蛋白序列比对(blastp)
如果要比对蛋白质序列(蛋白 vs 蛋白),使用blastp:
blastp -query query_protein.fa -db protein_db -out results.txt -evalue 1e-5 -outfmt 6 -num_threads 4
(3)DNA vs 蛋白比对(blastx)
如果你的查询序列是DNA但要比对蛋白数据库(先翻译再比对),使用blastx:
blastx -query query.fa -db protein_db -out results.txt -evalue 1e-5 -outfmt 6 -num_threads 4
这适用于**预测基因编码区(CDS)**的功能注释。
(4)蛋白 vs DNA比对(tblastn)
如果你的查询序列是蛋白,但目标数据库是DNA(将DNA翻译后比对),使用tblastn:
tblastn -query protein.fa -db genome_db -out results.txt -evalue 1e-5 -outfmt 6 -num_threads 4
这可以用于寻找基因组中的蛋白编码区。
(5)DNA vs DNA翻译比对(tblastx)
如果查询序列和数据库都是DNA,但希望都翻译为蛋白后再进行比对:
tblastx -query query.fa -db genome_db -out results.txt -evalue 1e-5 -outfmt 6 -num_threads 4
这种方法计算量较大,但可以检测远缘同源序列。
4. 结果筛选与分析
BLAST 结果通常需要进一步筛选:
(1)提取高质量比对结果
awk '$3 > 90 && $4 > 100' results.txt > filtered_results.txt解释:
$3 > 90:相似度 > 90%
$4 > 100:比对长度 > 100 bp
(2)统计比对数量
wc -l results.txt
(3)可视化BLAST比对
可以用blast_formatter转换为HTML格式:
blastn -query query.fa -db genome_db -out results.html -evalue 1e-5 -outfmt 7
然后用浏览器打开results.html查看。
5. 总结
完整BLAST流程
- 安装BLAST:
conda install -c bioconda blast - 准备数据库
:
- 下载NCBI数据库:
update_blastdb.pl --decompress nt - 自建数据库:
makeblastdb -in genome.fa -dbtype nucl -out genome_db
- 下载NCBI数据库:
- 运行BLAST比对:
blastn -query query.fa -db genome_db -out results.txt -evalue 1e-5 -outfmt 6 -num_threads 4 - 筛选结果:
awk '$3 > 90 && $4 > 100' results.txt > filtered_results.txt
生信大白记第58记,就到这里,关注我!
下一记,持续更新学习生物信息学的内容!
生信大白记邮箱账号:shengxindabaiji@163.com
生信大白记简书账号:生信大白记
生信大白记CSDN账号:生信大白记
生信大白记微信公众号:生信大白记
加入生信大白记交流群938339543

















 1552
1552

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









