






NCBI官网National Center for Biotechnology Information
https://www.ncbi.nlm.nih.gov/
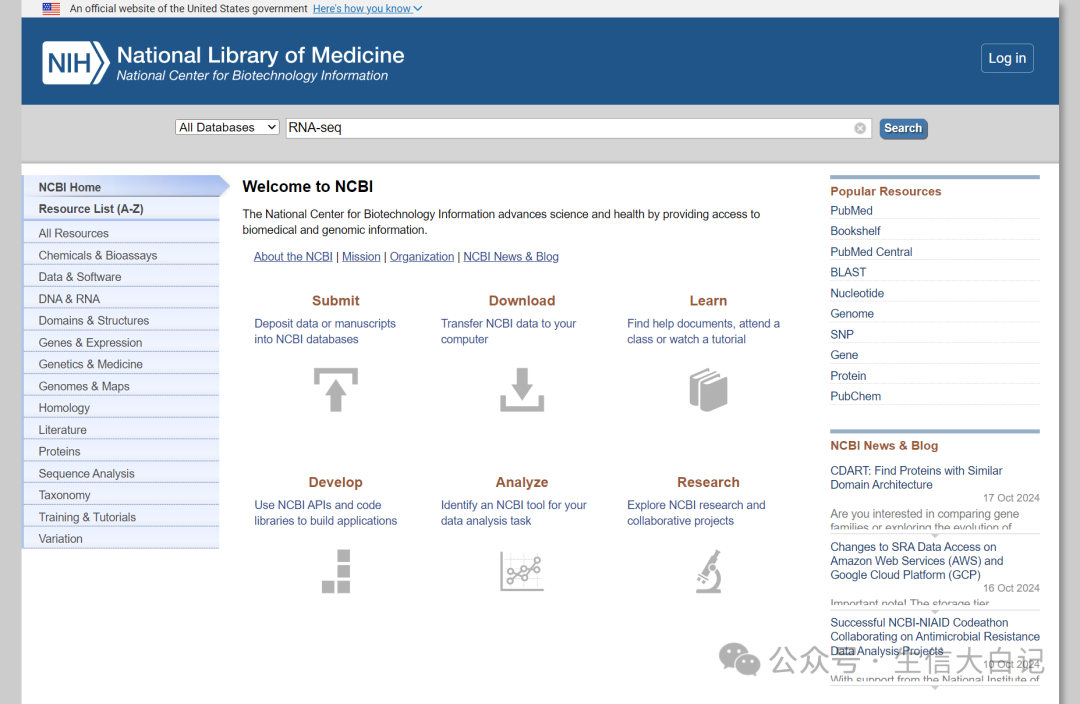
搜索RNA-seq, 进入GEO数据库
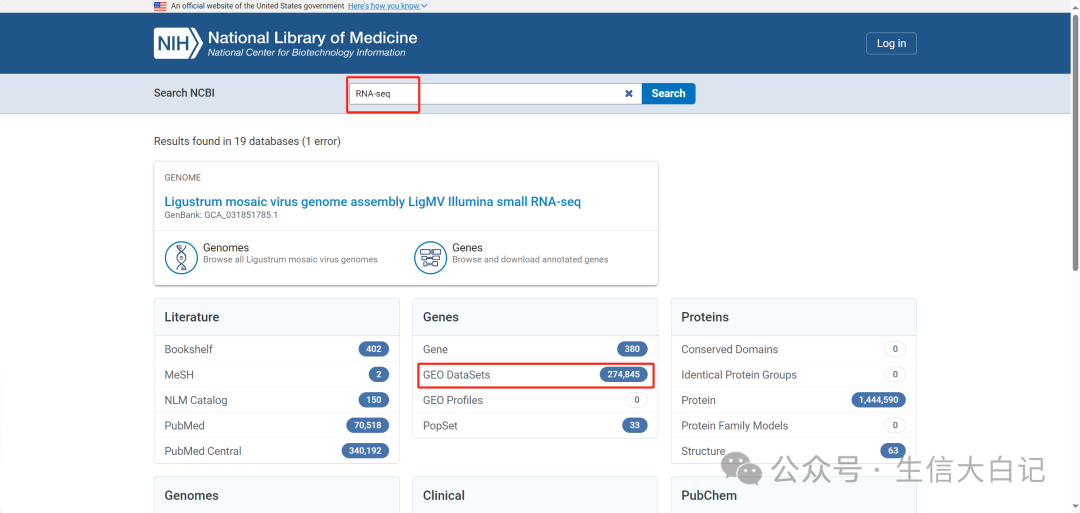
点击第一篇RNA-seq相关文章,作为案例
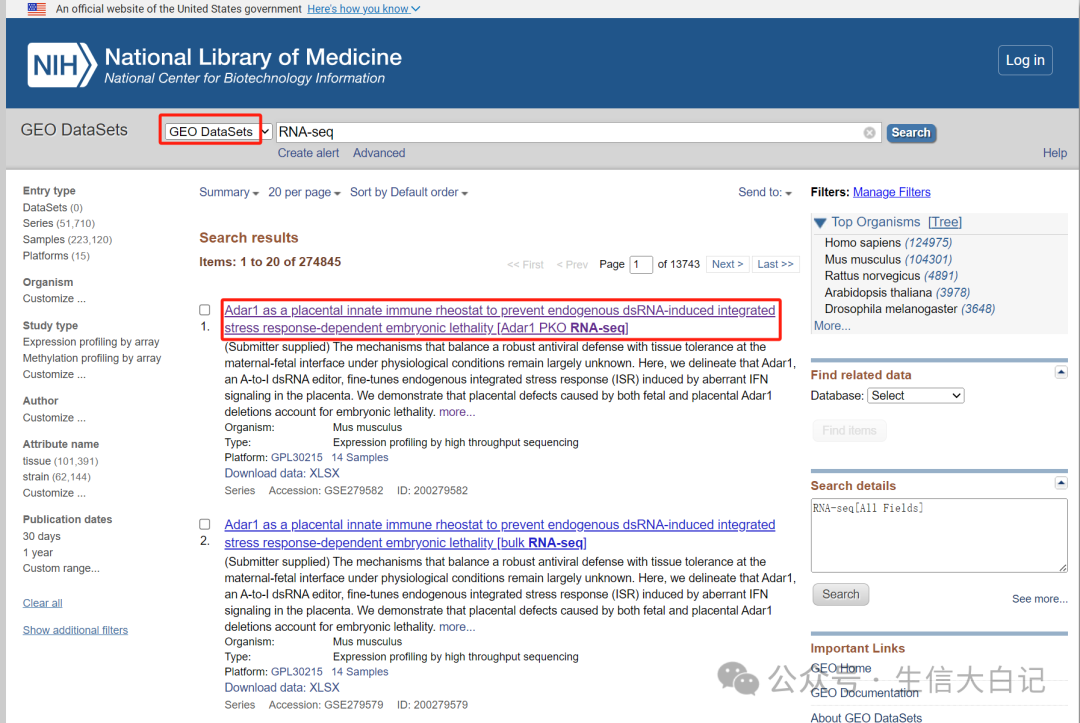
点击文章进去可以看到对应的GSE号
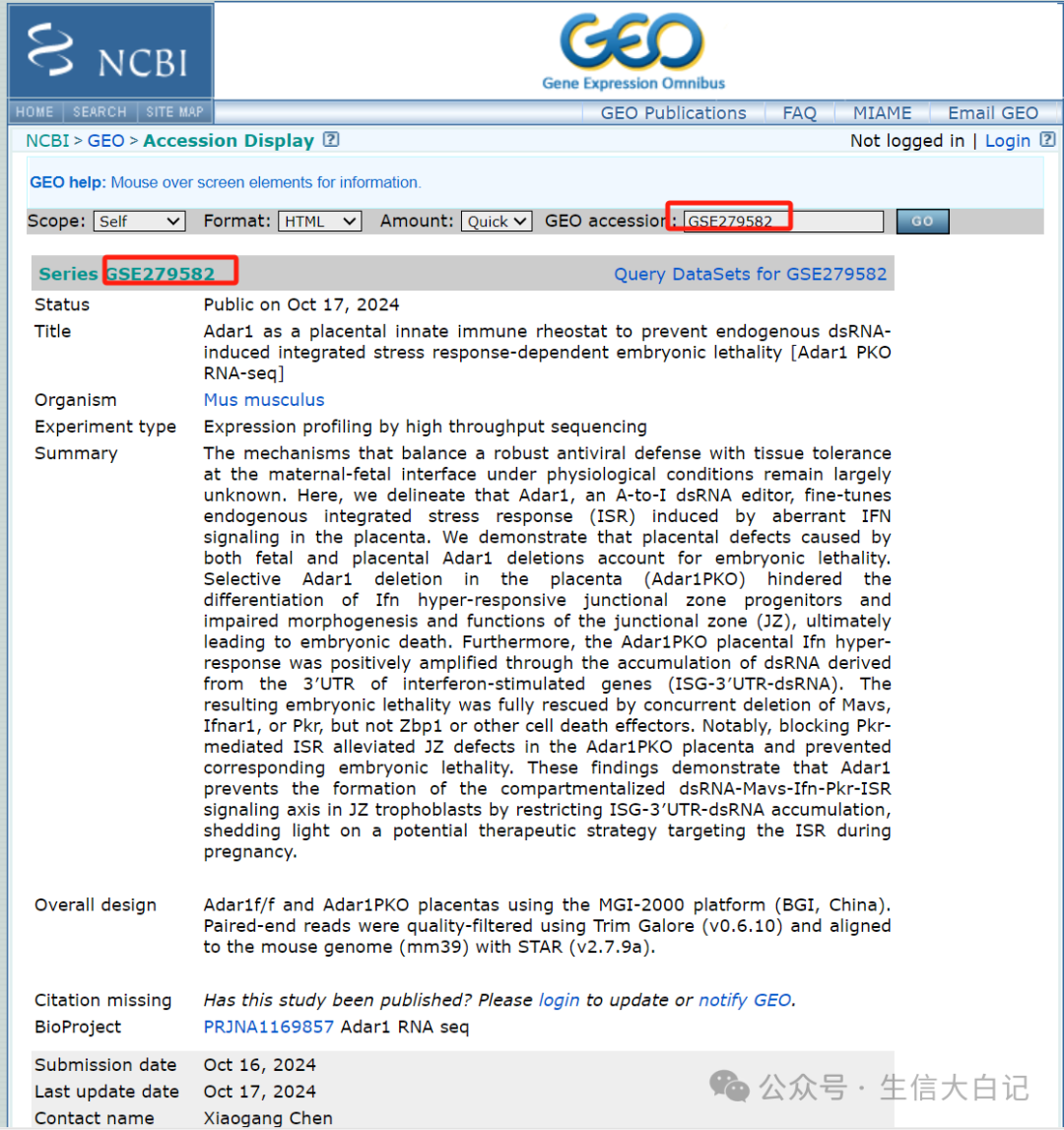
往下滑动可以看到对应的GSM号和SRA Run Selector
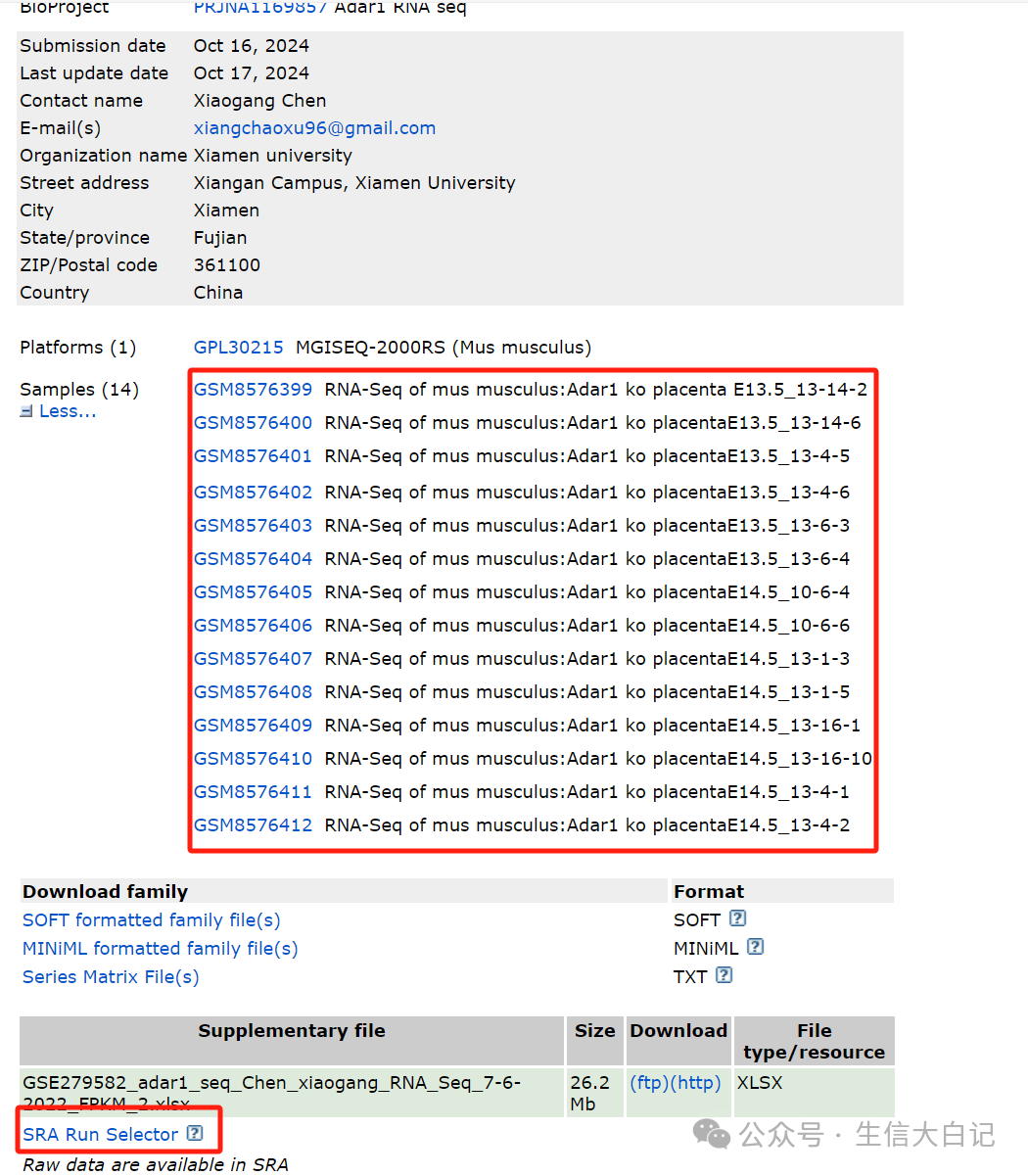
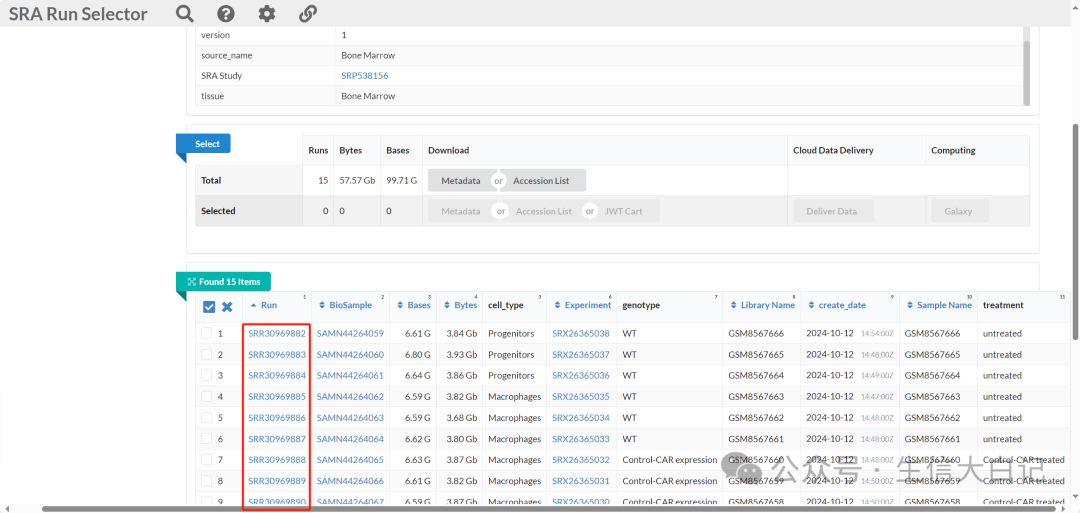
点击SRA Run Selector, 竟然没有对应SRR信息
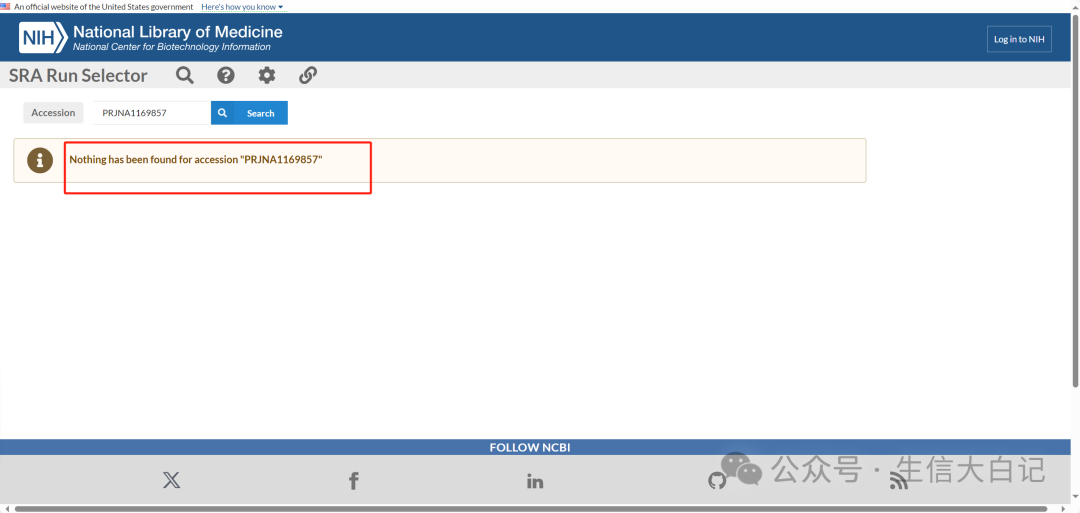
那就换一篇
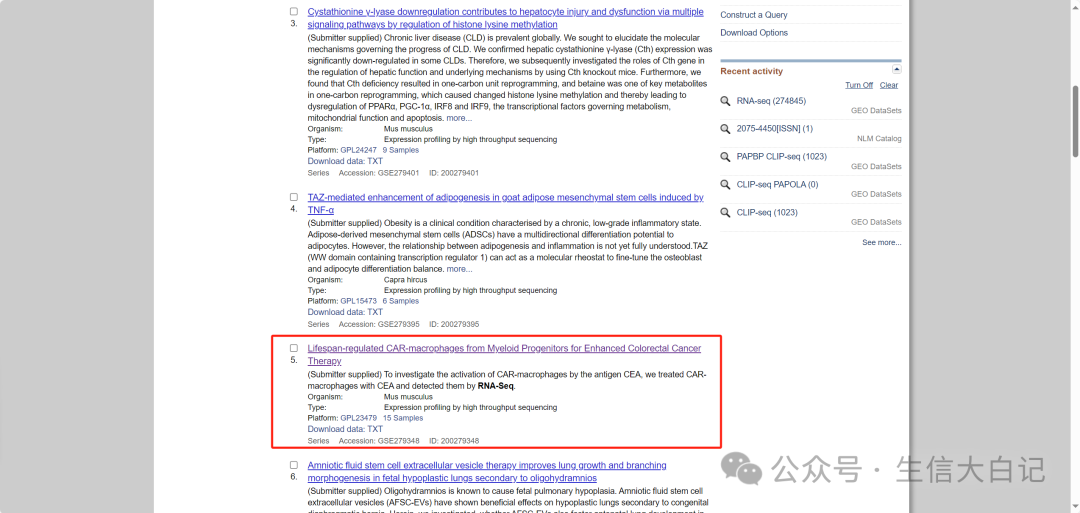
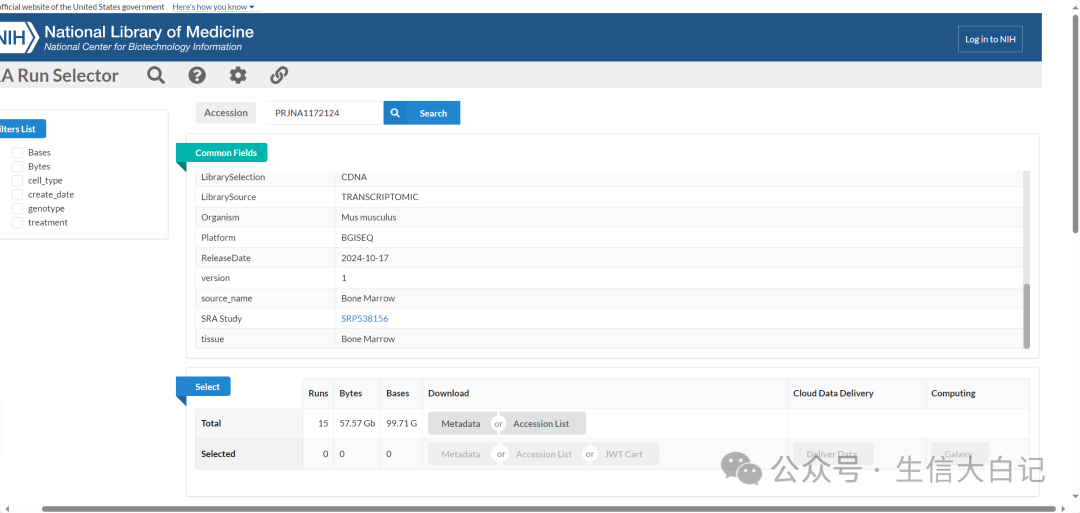
还是进入SRA Run Selector可以看到对应有SRR号,一个SRR号就是一个样本
在NCBI的数据库中,数据的组织层次结构比较复杂。以下是几个常见的编号(SRR、SRX、SRP等)及它们的含义:
1. SRR 号(SRA Run Number)
-
表示:一个具体的测序运行(Run)。
-
用途:指向一个具体的实验数据文件,通常是单个FASTQ文件或多个分割文件(如双端测序)。
-
例子:
SRR12345678 -
解析:每个SRR编号代表一次具体的测序过程及其输出的数据。下载时,SRR是最常用的编号。
2. SRX 号(SRA Experiment Number)
-
表示:一个实验(Experiment)。
-
用途:将多个测序运行(多个SRR)归属于一个实验。
-
例子:
SRX987654 -
解析:如果一个实验使用了不同的平台或多次重复测序,那么它会包含多个SRR编号。
3. SRP 号(SRA Project Number)
-
表示:一个项目(Study/Project)。
-
用途:将多个实验(SRX)归类为一个研究项目。
-
例子:
SRP234567 -
解析:SRP号通常对应于一个研究者提交的整体项目,用来描述一个研究计划的背景和目的。
4. PRJNA 号(Project Accession Number)
-
表示:NCBI BioProject编号,用于跟踪研究项目。
-
用途:PRJNA号是与SRP号类似的BioProject编号,但PRJNA可以跨数据库使用,包括基因组数据、RNA-Seq、和基因表达等不同类型的数据。
-
例子:
PRJNA123456 -
解析:它提供了项目的整体描述,并与SRA项目(SRP号)关联。
5. GSM 号(Gene Expression Sample)
-
表示:一个样品(Sample)。
-
用途:GSM编号用于Gene Expression Omnibus (GEO)数据库中的单个样品数据。
-
例子:
GSM345678 -
解析:GSM描述了某个具体生物样品的特性,并且可能包含RNA-Seq、微阵列数据等。
6. GSE 号(GEO Series)
-
表示:一个数据系列(Series)。
-
用途:将多个样品(GSM)组织为一个实验系列,表示实验的整体设计。
-
例子:
GSE678910 -
解析:GSE号描述了一个GEO研究项目中的实验设计,并将相关样品和数据进行分组。
7. SRA号(Sequence Read Archive Number)
-
表示:一个档案(Archive)。
-
用途:
SRA是整个序列数据存档数据库的名字,而SRA编号指向一个已提交的归档条目,通常是BioProject、实验、运行等信息的集合。 -
例子:
SRA123456 -
解析:这个编号用于访问整个项目或归档数据的一部分,但与SRP/PRJNA信息部分重叠。
现在开始下载数据
下载数据前得配置好sratoolkit
wget https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/2.11.3/sratoolkit.2.11.3-centos_linux64.tar.gztar -zxvf sratoolkit.2.11.3-centos_linux64.tar.gz #解压vi ~/.bashrc #用vi编辑器编辑bashrc文件i #由command line 进入insertion lineexport PATH=$PATH:/data1/ganyuli/up_download_tool/sratoolkit.2.11.3-centos_linux64/bin #这里是bin文件的绝对路径Esc :wq #退出并保存source ~/.bashrc #让配置生效cd /data1/ganyuli/up_download_tool/sratoolkit.2.11.3-centos_linux64/bin./vdb-config --interactive #执行vdb-config --interactive
配置到这还没完,得进入图形界面进行操作后,后面才能运行程序
cd /data1/ganyuli/up_download_tool/sratoolkit.2.11.3-centos_linux64/bin
./vdb-config --interactive #运行这条命令后就会进入图形界面
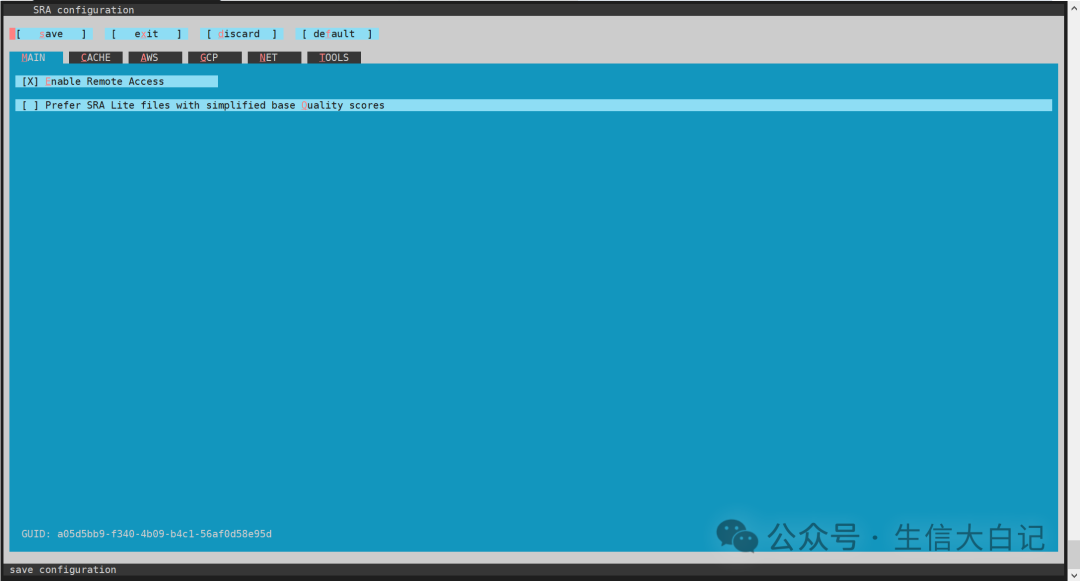
Tab键移动到CACHE,然后enter键进入
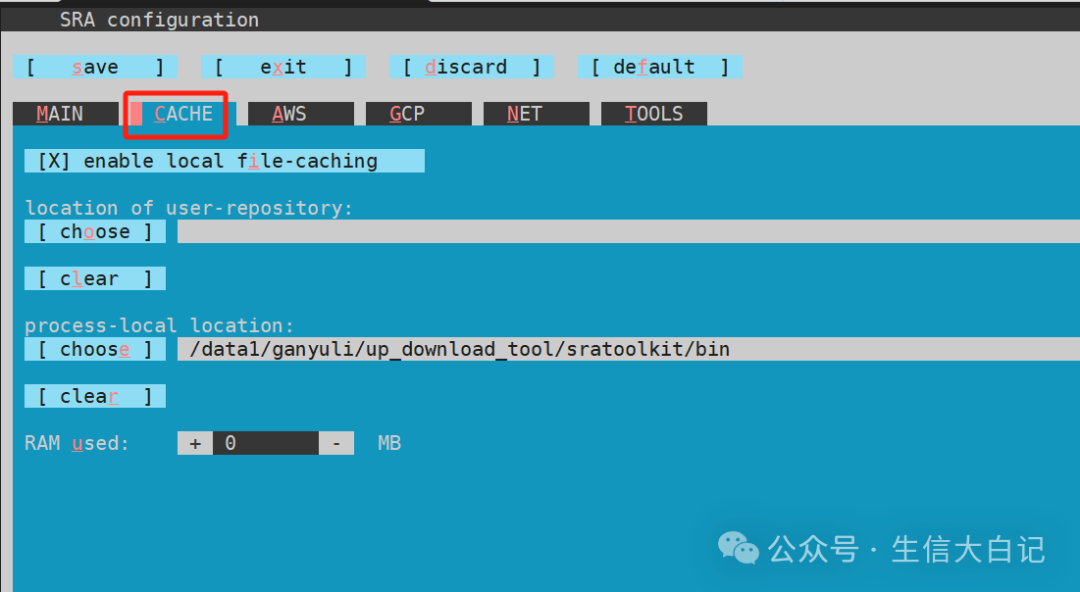
Tab键移动到choose
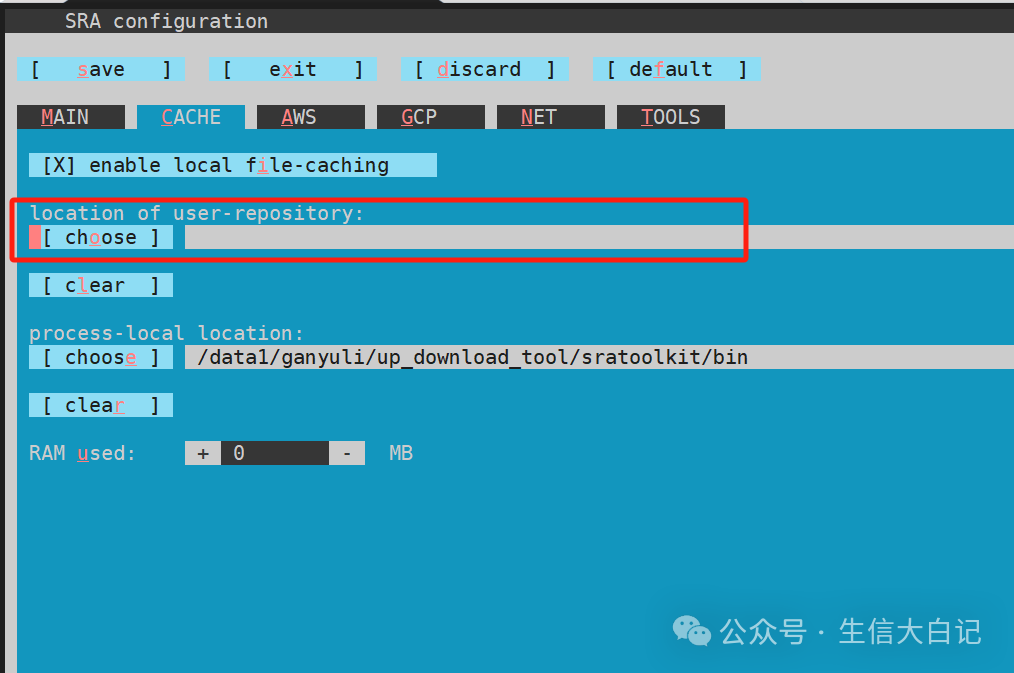
enter键进入chose, 选择一个默认的数据下载具体路径,到时候程序命令下载如果不指定输出路径,就会默认输出到这个路径
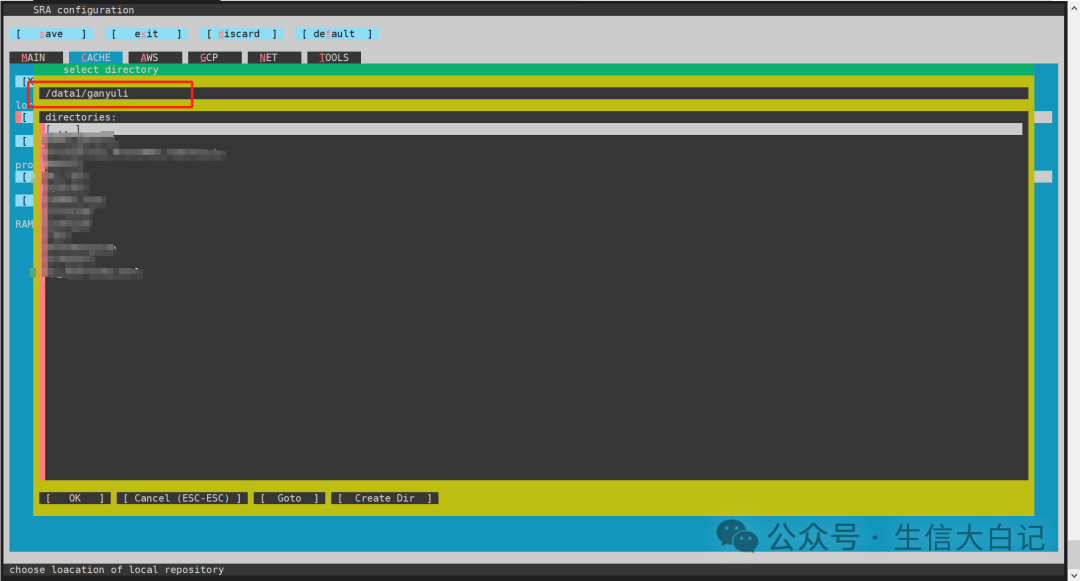
Tab移动到OK处,enter回车,选择yes再回车,再ok
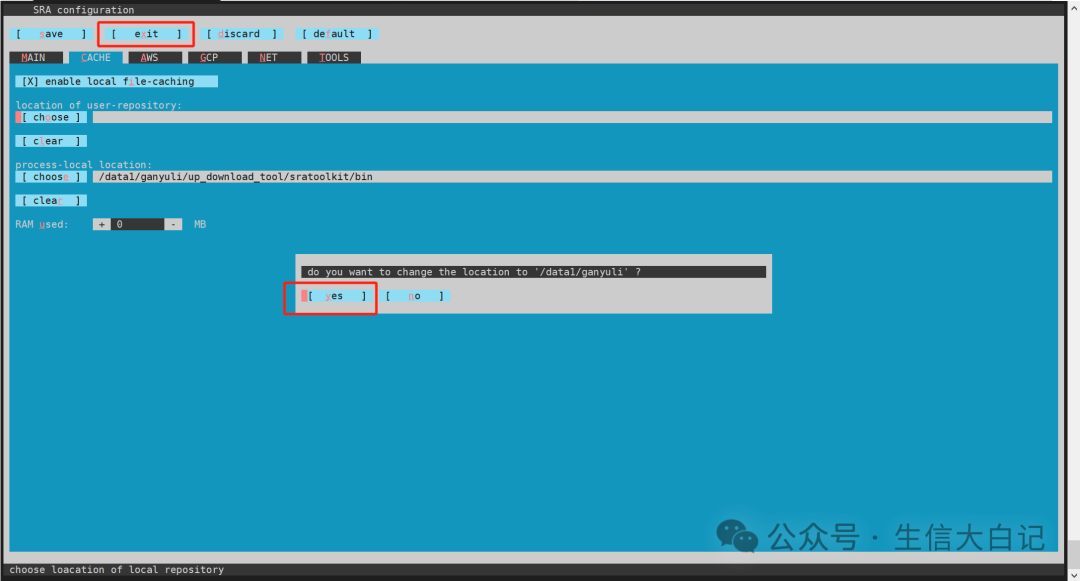
最后exit, yes, ok
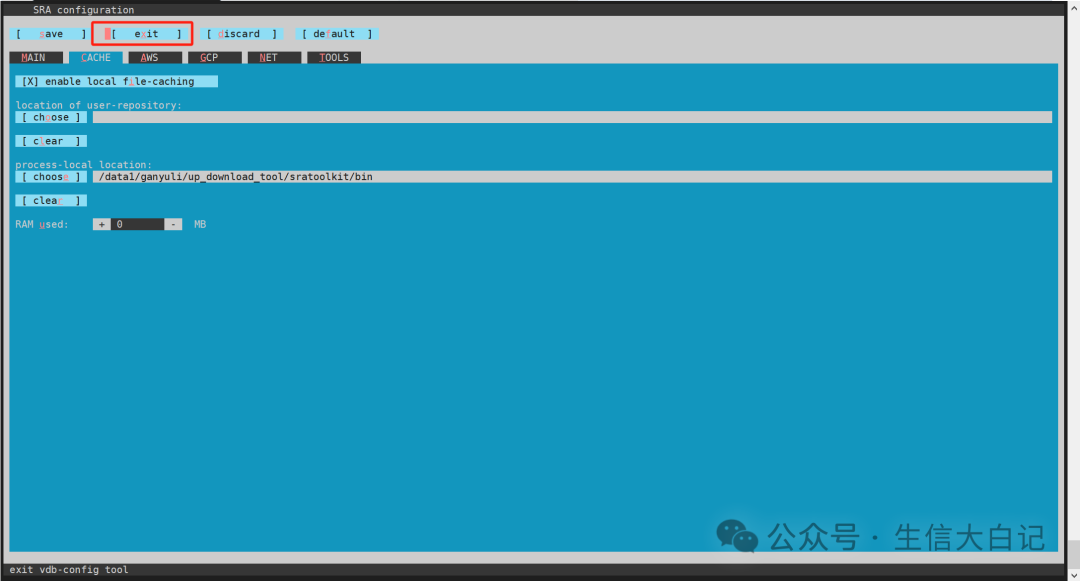
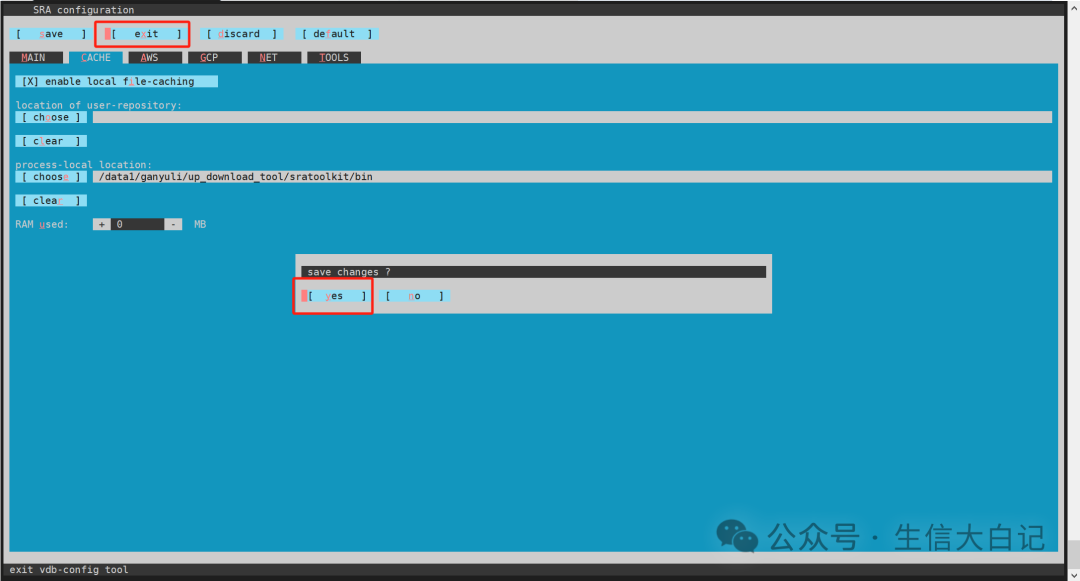
这样就配置完成了
数据下载命令很简单
#单个数据下载,输出到当前目录/data1/ganyuli/up_download_tool/sratoolkit/bin/prefetch SRR30969882 -O ./# 下载后数据格式是sra格式,转换这个数据的格式为fastq,输出到当前目录# 下面这个命令的参数,可以不用管数据是单端还是双端,是自动识别的,转换出来是是啥就是啥/data1/ganyuli/up_download_tool/sratoolkit/bin/fastq-dump --split-3 --gzip ./SRR30969882/SRR30969882.sra -O ./# 批量下载# 可以准备一个SRR.txt文件,里面只有一列,每行都是一个SRR号,并且保证是Unix格式for i in `cat SRR.txt`doif [ -e "fastq/${i}_2.fastq.gz" ]; thenecho "$i has been dowloaded"else/data1/ganyuli/up_download_tool/sratoolkit/bin/prefetch $i -O ./sra /data1/ganyuli/up_download_tool/sratoolkit/bin/fastq-dump --split-3 --gzip ./sra/$i/$i.sra -O ./fastq fi done# 如果转换格式太慢,可以换成fasterq-dump,快一点fasterq-dump SRR30969882.sra
生信大白记第17记,就到这里,关注我!
下一记,持续更新学习生物信息学的内容!
生信大白记邮箱账号:shengxindabaiji@163.com
生信大白记简书账号:生信大白记
生信大白记CSDN账号:生信大白记
生信大白记微信公众号:生信大白记
加入生信大白记交流群938339543

















 1459
1459

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









