







circlize包画圈到底怎么画?以下我将以笔记的方式记录我的学习过程
方法详见:Circular Visualization in R
我先看了官方文档,看了两天实在是没看懂,然后看的下面这篇博客,感谢大佬 在R中利用circlize绘制圈图_生信菜鸟1号的博客-CSDN博客 https://blog.csdn.net/weixin_45694863/article/details/129299114
https://blog.csdn.net/weixin_45694863/article/details/129299114
目录
2.2. function(region,value)类型的绘图
1. 绘图前准备
1.1 安装包以及圈图参数
rm(list = ls())
#如果没有安装过circlize包,则把下面这行的"#"去掉
#install.packages("circlize")
library(circlize)
#重置画圈图的参数
circos.clear()
#参数
circos.par("track.height"=0.1,gap.degree=3, start.degree =90,
clock.wise = T,track.margin=c(0,0.02), cell.padding=c(0,0,0,0))
- start.degree 绘图起始位置,默认从水平方向开始(3点钟方向),90表示12点钟
- gap.degree track左右间隔大小,gap.after等价
- track.margin track上下间隔大小 c(,)
- track.height 每个track的默认高度,和track.margin单位一样,都是半径的百分比
- cell.padding 看不懂,默认吧 c(,)
- clock.wise 排列方式,顺时针 T/F
- xaxis.clock.wise x轴,顺时针 T/F
1.2 准备数据
circos.initializeWithIdeogram(species = "hg19" #物种mm10,hg19
#chromosome.index=c(paste0('chr',1:19),'chrX')
#选择染色体,默认全部,我们只用到1-19和x
)
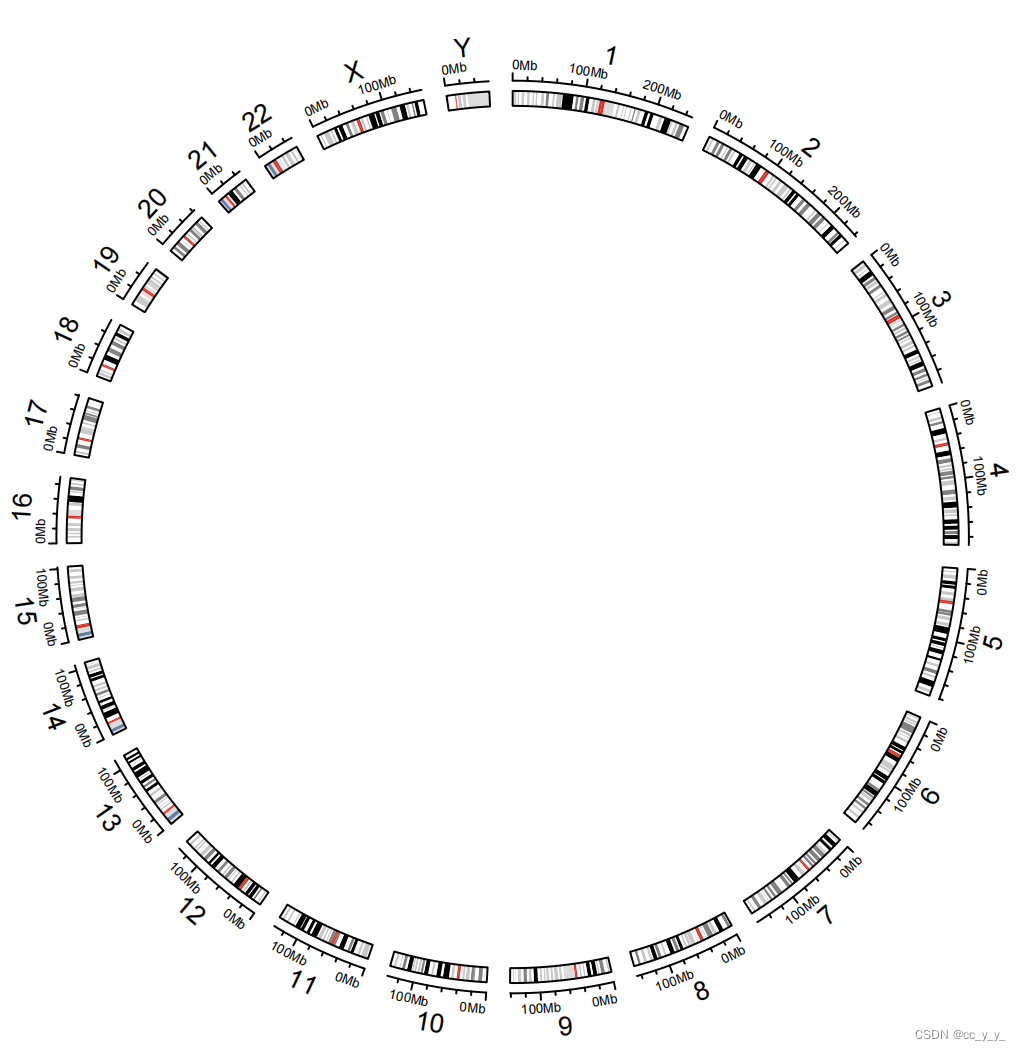

使用 circos.initializeWithIdeogram() 初始化圆形图,会创建两个轨迹,一个用于包含轴和染色体名称,另一个轨迹用于绘制Ideogram(染色体G带图) ;
G带是什么?G带(Giemsa-banding)将染色体制片经盐溶液、胰酶或碱处理, 再用吉母萨染料染色, 在光镜下进行检查, 见到特征性的带。一般富含AT碱基的DNA区段表现为暗带,富含GC碱基的DNA区段表现为亮带。
circos.initializeWithIdeogram() 默认使用的是 hg19 版的 cytoband 数据,可以使用 species 参数来指定为 hg18 或其他物种,cytoband 数据格式如下,band是cytogenetic带的名称,stain列表示吉姆萨氏染色的结果(没看懂,不管了,这两圈不重要,先学圈图怎么画)
chrom start stop band stain
chr1 0 2300000 p36.33 gneg
chr1 2300000 5400000 p36.32 gpos25
chr1 5400000 7200000 p36.31 gneg
chr1 7200000 9200000 p36.23 gpos25
chr1 9200000 12700000 p36.22 gneg
chr1 12700000 16200000 p36.21 gpos50
chr1 16200000 20400000 p36.13 gneg
chr1 20400000 23900000 p36.12 gpos25
chr1 23900000 28000000 p36.11 gneg
chr1 28000000 30200000 p35.3 gpos25
chr1 30200000 32400000 p35.2 gneg然后就是绘图所需的数据了,包里有自带的示例数据,可以看到顺序就是chr start end value
bed = generateRandomBed(nr = 200) 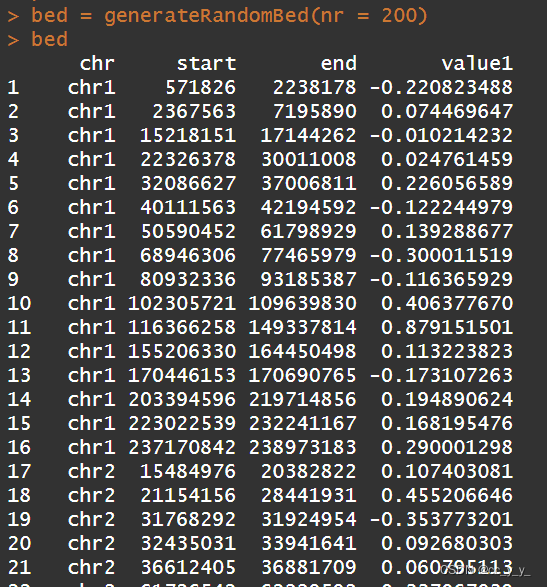
绘制不同图形用不同数据,比如绘制线图点图有chr start value就好,如果绘制heatmap或者genomicRect这样的范围图则chr start end value都需要。
2. 绘图
2.1 function(x,y)类型的绘图
function(x,y)类型的绘图包括折线图,点图等
2.1.1 折线图
方法1:在track中加入circos.lines绘制折线图
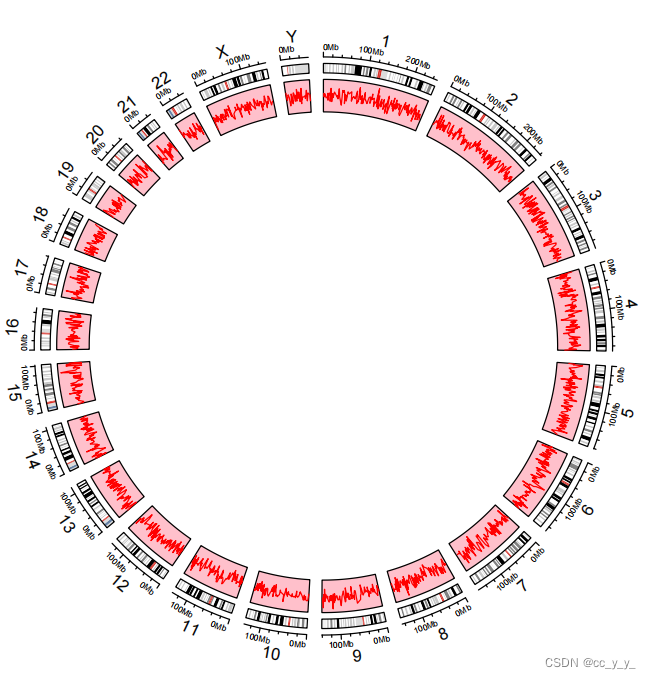
#绘图就用circos.track,这个函数表示新建一个层,需要参数:染色体,x,y。里边circos.lines用于在层中绘制折线图。
circos.track(factors=bed$chr, x = bed$start, y = bed$value1, track.index=3,
track.height = 0.1,ylim=c(-2,2),bg.col="pink",
panel.fun = function(x, y) {
circos.lines(x, y, col = "red") #默认折线图,x和y来自上边的参数传递
})
具体track里边的参数如下:
- factors 染色体列
- x 一般start列
- y 一般value列,一般只有折线图点图之类的需要xy,其他都是具体区域#factors,x和y有时候不需要设置,因为有的绘图需要的是region和value的格式
- track.index track索引,定义绘制在哪个track里,从外到内数,我们红色的这一层是第三层
- track.height 高度,在circos.par可以定义默认的,也可以在track里自定义,指层高,即这一层圆环有多宽(按占整个圈图的百分比算,0.1表示层高是圈图半径的10%)
- ylim指y坐标轴的最大值和最小值(范围)
- bg.col = NA, track的背景色,可以是一个也可以是染色体数目个
- bg.border = “black” track的边框,可以是NA
后面几个不懂:
- bg.lty = linetype
- bg.lwd =linewidth
- panel.fun 绘图函数 function(x,y) function(region,value)
- track.margin only affect current track
- cell.padding only affect current track
circos.track函数相当于进行一个For循环,每次绘制一条染色体,所以在function内部可以用CELL_META$sector.index 获取每次绘制时当前染色体的index,用CELL_META$ylim 获取y的范围,利用用CELL_META$xlim 获取x的范围,利用这一特点我们还可以为折线图绘制阈值线。加阈值线的panel.fun中使用circos.segments。
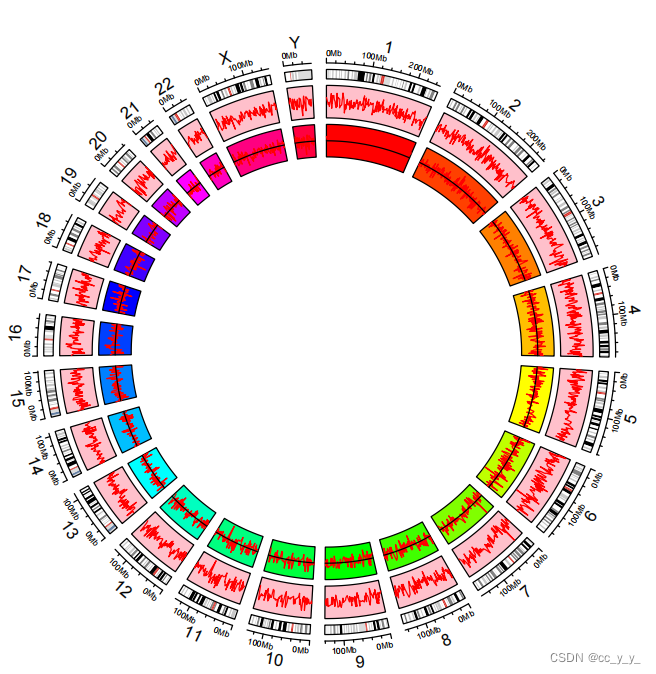
th=quantile(bed$value1,0.5)
circos.track(factors=bed$chr, x = bed$start, y = bed$value1,#track.index=3,
bg.col=rainbow(24),
track.height = 0.1,ylim=c(-2,2),
panel.fun = function(x, y) {
circos.lines(x, y, col = "red") #默认折线图
circos.segments(x0=CELL_META$xlim[1],x1=CELL_META$xlim[2], #添加segment
y0=th,y1=th,col = 'black')
})
})
注意:这里没有加track.index,如果不定义track的index,多次运行这个circos.track,绘制会依次绘制。而如果加了track.index,多次运行这个circos.track,就会在同一层多次画,且之前画的不会清除。
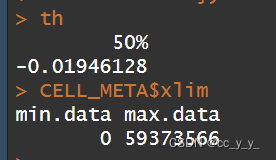
上述代码中,th是什么?CELL_META$xlim又是怎样的?
th是value1这一列的值从小到大排列,排在50%处的值,CELL_META$xlim是x的范围
方法2:
先绘制一个空的track,再用circos.trackLines绘制折线图
circos.track(ylim=c(-2,2),track.height = 0.1)
circos.trackLines(factors =bed$chr,x=bed$start,y=bed$value1,col='red')2.1.2 点图
circos.track(factors=bed$chr, x = bed$start, y = bed$value1,#track.index=3,
#bg.col=rainbow(19),
track.height = 0.1,ylim=c(-2,2),
panel.fun = function(x, y) {
circos.points(x, y, col = "red",cex =0.3)
circos.segments(x0=CELL_META$xlim[1],x1=CELL_META$xlim[2],
y0=th,y1=th,col = 'black')
}) 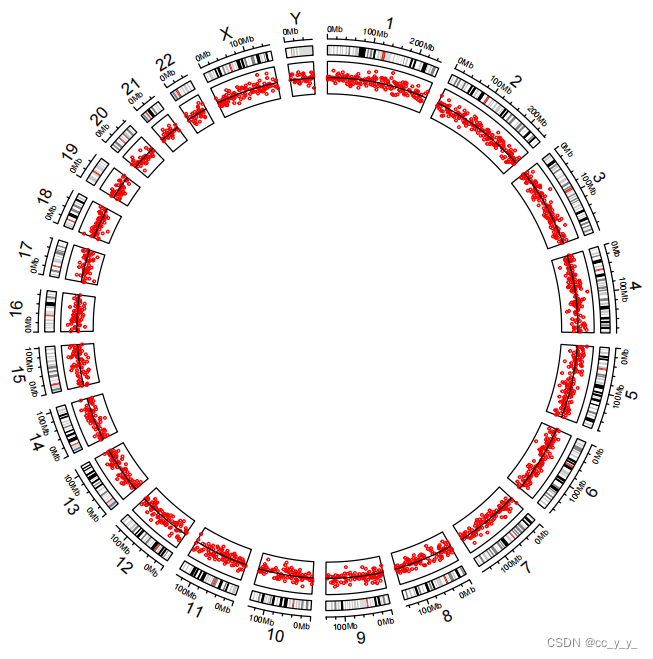
与折线图的区别在于,一个用的是circos.lines(),一个用的是circos.points
cex可以用来调整点的大小
2.2. function(region,value)类型的绘图
这种绘图需要的数据是chr start end value,是定义某个范围内的数值大小
2.2.1.circos.genomicRect
circos.genomicTrack(bed,ylim=c(0,1), #bed是数据名,不用设置chr,x,y了
panel.fun = function(region, value, ...) { #resion就是 chr start end value就是value,排列顺序对了就ok
circos.genomicRect(region, value, ytop= 1, ybottom = 0,
#ytop.column 使用第几列value,或者自定义ytop
col = 'transparent',border='#2686e3',lwd=0.4)
circos.lines(CELL_META$cell.xlim, c(0.5, 0.5), lty = 2, col = "red")
}, stack = F,track.height = 0.1)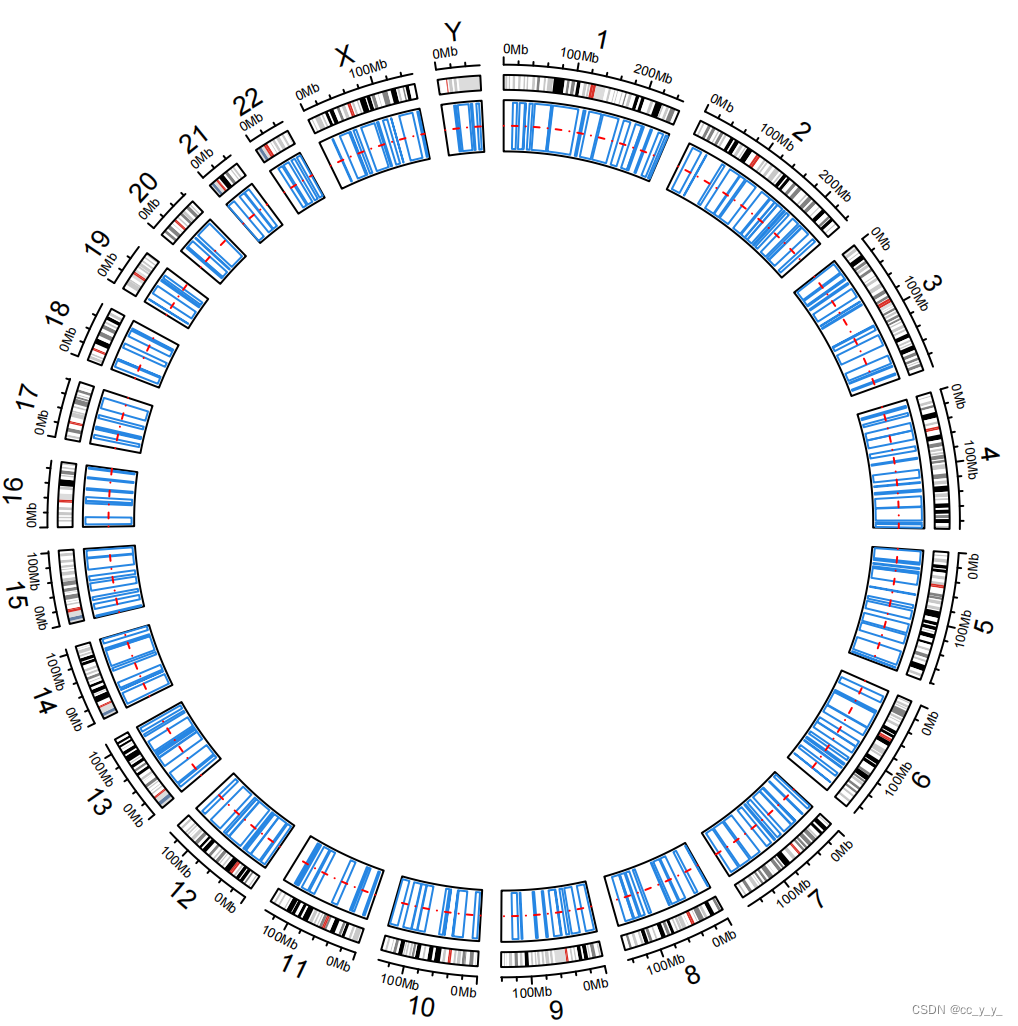
- 每个区间都是x的坐标,定义ytop和ybottom后,每一行数据就围成了一个矩形,中间是空的,举行的边界线就是这些蓝色的线。
- stack = F不知道啥意思,如果定义为T,蓝色矩形会一半在track内一半在track外。
- circos.lines在y=0.5处画了一条红色的虚线
3. 外圈的染色体如何画
需要提供包含染色体名称与长度对应的文件,例如:
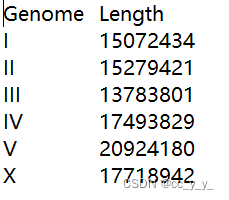
读文件,然后初始化
ref<-read.table("C:\\Users\\admin\\Desktop\\chr_len.txt", header = TRUE)
circos.initialize(factors=ref$Genome,
xlim=matrix(c(rep(0,6),ref$Length),ncol=2))
circos.track(ylim=c(0,1))
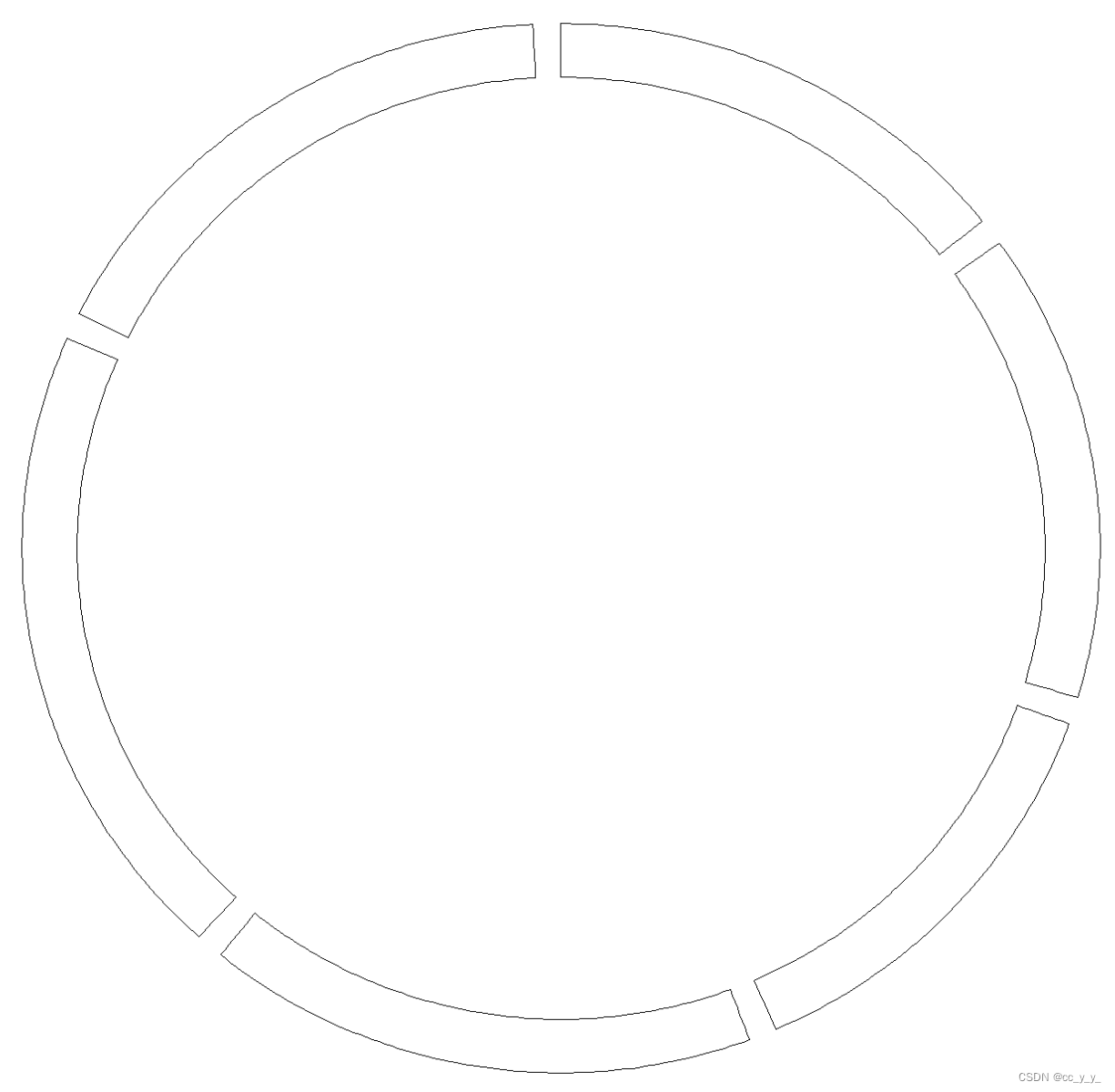
加上刻度标签和染色体名称
#设置染色体框
#circos.track每条染色体按顺序轮流function,相当于一个for(chr1:chrn)循环,track.index表示绘图track的索引
circos.track(ylim=c(0,1),panel.fun=function(x,y) {
Genome=CELL_META$sector.index #染色体ID
xlim=CELL_META$xlim #染色体长度范围
ylim=CELL_META$ylim #y的范围,上面设置的是0-1,所以这里是0-1
#circos.rect(xlim[1], 0, xlim[2], 1, col = rand_color(19)) #跟bg.col等价
circos.text(mean(xlim),mean(ylim),Genome,cex=0.8,col="white",font = 2,
facing = "inside", niceFacing = TRUE)
},bg.col= rand_color(20),bg.border=F,track.height=0.1)
brk <- seq(0,200,2)*10^6
#设置x轴axis
#track.index = get.current.track.index() 可以让轴外放
circos.track(track.index = get.current.track.index(), panel.fun = function(x, y) {
circos.axis(h="top",major.at=brk,labels=round(brk/10^6,1),labels.cex=0.4,
labels.pos.adjust =0,
col='#045eaf',labels.col='#045eaf',lwd=0.7,labels.facing="clockwise")
},bg.border=F) 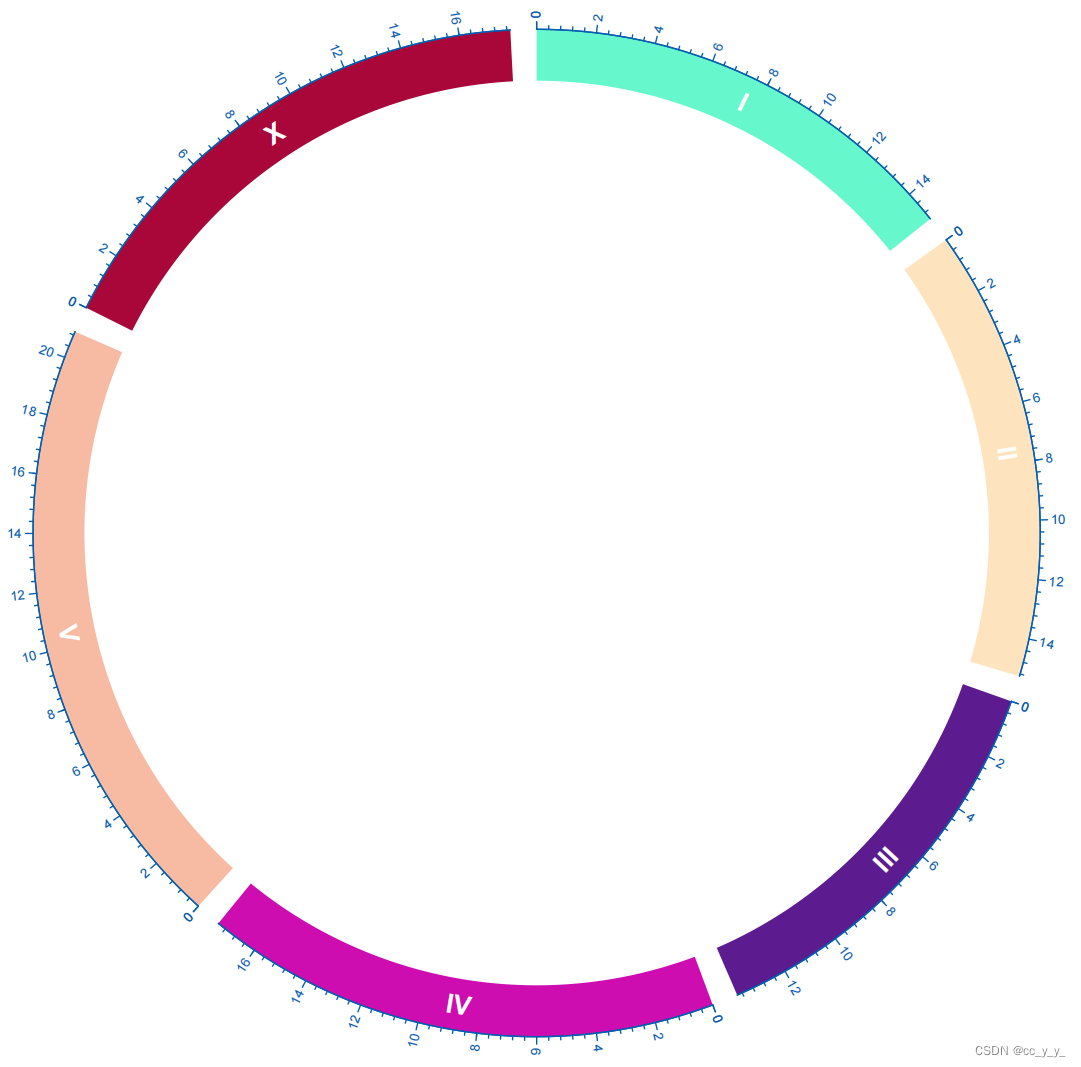
ps:记录遇到过的报错:
我们画圈图一般都是对表格文件中的某几列数据可视化统计,表格文件导入R中使用 ,读入为dataframe类型,当dataframe中含有字符类型的数据时,注意在数据框建立时加入语句:
stringsAsFactors = FALSE
当我们在建立数据框的时候,R语言将会默认把字符型(character)当成因子(factor),factor数据类型不能像character类型一样切片

















 1774
1774

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









