







Yunhe Wang, Zhuohan Yu, Shaochuan Li, Chuang Bian, Yanchun Liang, Ka-Chun Wong, Xiangtao Li, scBGEDA: deep single-cell clustering analysis via a dual denoising autoencoder with bipartite graph ensemble clustering, Bioinformatics, Volume 39, Issue 2, February 2023, btad075
论文地址
代码
动机
单细胞RNA测序(scRNA-seq)是一种日益流行的技术,用于在单个细胞水平上对基因表达进行转录组分析。细胞类型聚类是对scRNA-seq数据进行分析的首要任务,有助于准确识别细胞类型并研究其转录本的特征。最近,基于深度自动编码器和集成聚类的几种计算模型已被开发用于分析scRNA-seq数据。然而,当前的深度自动编码器不足以学习scRNA-seq数据的潜在表示,并且从这些特征表示中获得一致的分割仍未得到充分探索。
为了解决这一挑战,作者提出了一种称为scBGEDA的单细胞深度聚类模型,通过双重去噪自动编码器和二分图集成聚类来识别单细胞转录组文件中的特定细胞群体。首先,提出了一种单细胞双重去噪自动编码器网络,将数据投影到压缩的低维空间,并通过明确建模零膨胀负二项重建损失和去噪重建损失的协同优化来学习特征表示。然后,设计了一种二分图集成聚类算法,通过基于图的一致性函数利用细胞之间的关系和学习的潜在嵌入空间。使用多种聚类度量在来自不同测序平台的20个scRNA-seq数据集上进行了多次比较实验。实验结果表明,scBGEDA在这些数据集上优于其他最先进的方法,并且还展示了其对大规模scRNA-seq数据集的可伸缩性。此外,scBGEDA能够识别细胞类型特定的标记基因,并通过量化基因对细胞聚类的影响进行功能基因组分析,从不同角度带来了对识别细胞类型和表征scRNA-seq数据的新见解。
模型

scBGEDA框架包括三个组件,包括数据处理步骤来建模高维度的scRNA-seq数据,一个单细胞双重去噪自动编码器网络和一个二分图集成聚类算法(图1)。作者提出了一个单细胞双重去噪自动编码器,将ZINB模型融入到去噪自动编码器网络中,以更好地捕捉scRNA-seq数据的结构。第二个模块的编码器接受经过数据过滤和归一化处理的预处理基因表达矩阵。通过主解码器和从解码器重构scRNA-seq数据的潜在表示。然后,作者设计了一个基于二分图和启发自集成聚类的传输切割方法的scBGEDA中的二分图集成聚类方法(Huang等,2019)。它包括两个阶段,第一个阶段通过K均值聚类方法生成一组基本的聚类;在第二个阶段,产生一个采用样本和聚类作为图节点的二分图,通过整合多个基本聚类来执行一致性函数。最后,通过解决广义特征值问题提供一致性聚类结果。
单细胞双重去噪自动编码器网络
单细胞双重去噪自动编码器网络基于ZINB模型学习scRNA-seq数据的潜在特征表示。它通过编码器和解码器中的堆叠层来捕获scRNA-seq表达矩阵的表示嵌入。编码器用于将scRNA-seq数据矩阵X映射到低维潜在特征表示Z,从输入中提取唯一信息。Z的维度远小于X的维度,以避免“维度诅咒”(Xie等,2016)。为了防止深度学习中的过拟合现象,输入的scRNA-seq数据被随机高斯噪声破坏,然后,自动编码器由全连接层构建。因此,编码器的映射函数可以定义为:

其中,X是输入的scRNA-seq表达矩阵,e是可以合并到编码器每一层的随机高斯噪声,Xcorrupt是输入数据的损坏数据,f W是编码器函数,W是函数的可学习权重,Z是编码器的输出特征表示向量。
解码器将潜在特征表示Z作为输入,旨在从低维特征表示Z中重构输入。由于重构和聚类任务之间不可避免的权衡,重构损失通常是聚类的次要优化目标。通常,重构损失主要由潜在空间的分布和解码器的重构能力确定。然而,在聚类过程中解码器网络的重构能力是不必要的。为了为scRNA-seq数据的聚类分配生成更具辨别力的特征,作者基于ZINB构建了一个跟随解码器来近似主解码器。双重去噪自动编码器网络的解码器可以定义为:
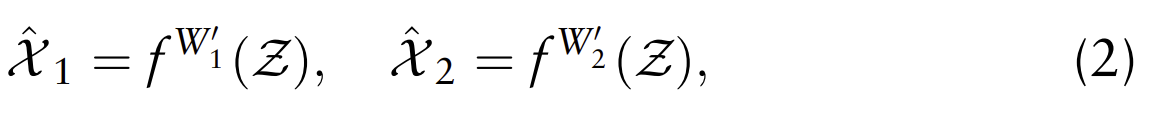
其中,f W1和f W2是主解码器和跟随解码器的函数,W1和W2表示权重参数矩阵,X^1和X^2分别是两个解码器的输入的重构。为了捕获scRNA-seq数据的特征,主解码器采用基于ZINB的自动编码器模型损失来表征原始计数数据。具体来说,ZINB用于基于零部件和负二项分布的组合数学建模scRNA-seq数据中的丢失事件,可以定义为:
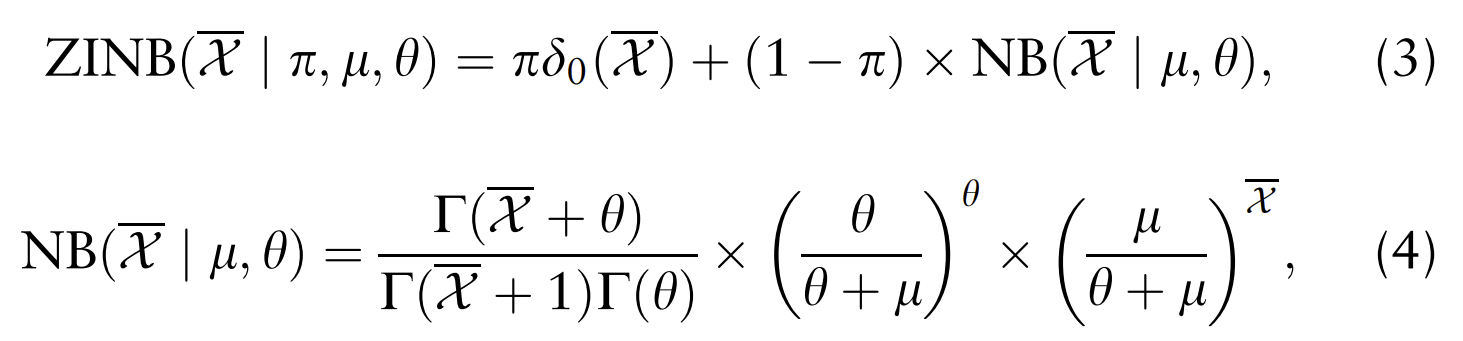
为了对ZINB分布进行建模,解码器网络具有三个输出层来计算三组参数。估计的参数可以定义为:
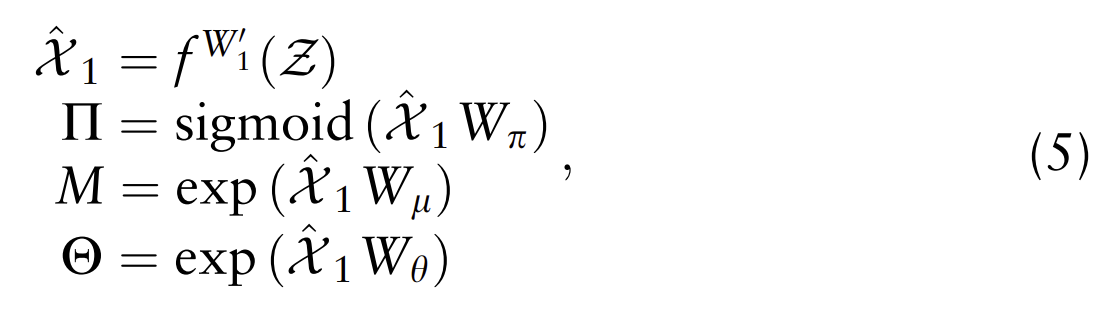
主解码器的重构损失函数采用ZINB似然的负对数,可以表示为:
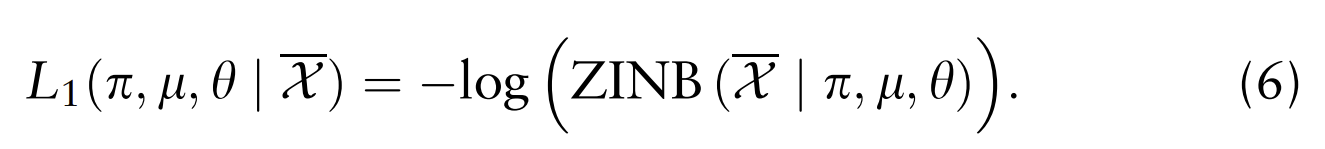
对于跟随解码器,提议通过将潜在表示Z传输到重构ZINB模型损失中的均值l参数来近似主解码器。通过排除,使这种双重去噪自动编码器模型更加健壮。因此,跟随解码器的损失可以写成:
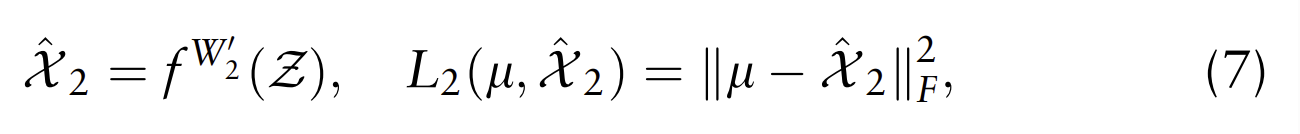
这是常规的MSE损失函数,并采用ReLU函数作为激活函数。为了保证潜在空间中特征表示的质量,将MSE损失添加到原始重构损失中,产生双重重构损失来学习解码器网络。双重去噪自动编码器的学习过程旨在通过最小化双目标损失函数来训练模型,可以定义为:
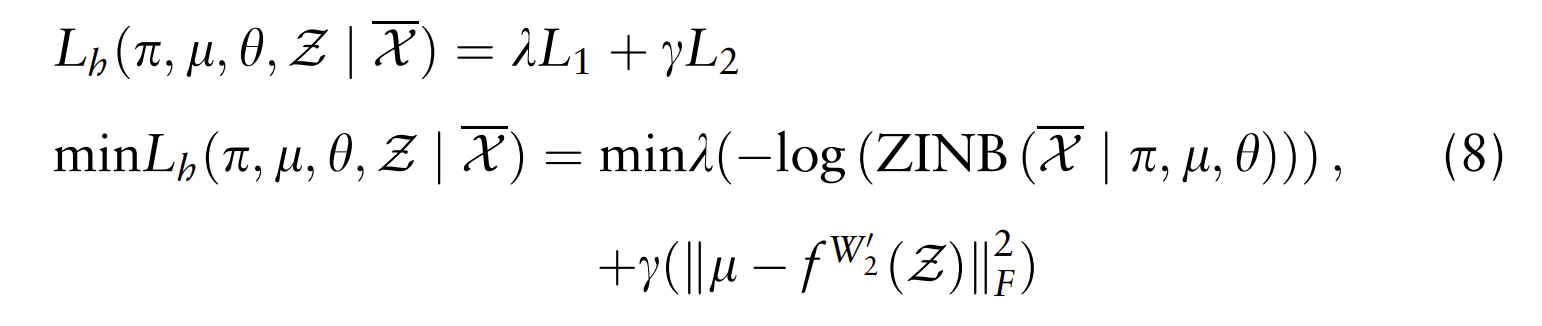
基本聚类生成
获得单细胞RNA测序数据表示Z = {z1, z2, ..., zn},其中n个样本来自潜在空间后,模型scBGEDA旨在利用Z中样本之间的关系,并通过二分图集成聚类来识别scRNA-seq数据的细胞类型。首先,为了确保对scRNA-seq数据集进行快速聚类,采用K均值聚类算法生成一组基本聚类,可以表示为:
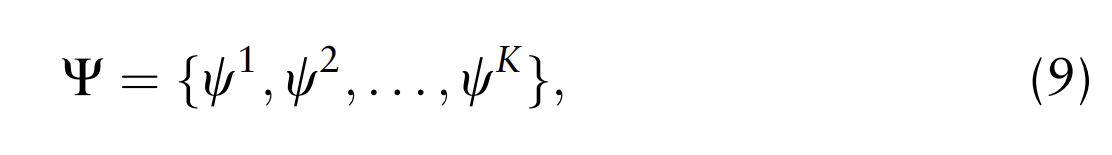
聚类数ki是从kmin到kmax中随机选择的整数,其中kmin和kmax分别表示聚类数的下限和上限。
二分图生成
为了获得稳健的一致性聚类结果,采用样本和聚类作为图节点(Huang等,2019),一个二分图G可以定义如下:

其中Z是特征表示;![]() 是聚类集合,并可以表示为:
是聚类集合,并可以表示为:
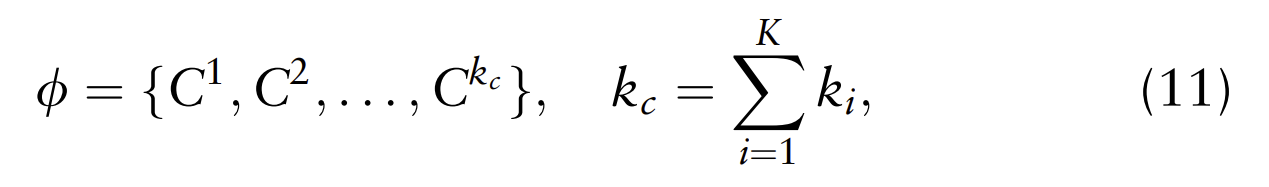
其中Ci是第i个聚类,ki是基本聚类中的聚类数,kc是聚类总数。此外,B代表反映Z和![]() 之间关系的交叉亲和矩阵,定义如下:
之间关系的交叉亲和矩阵,定义如下:
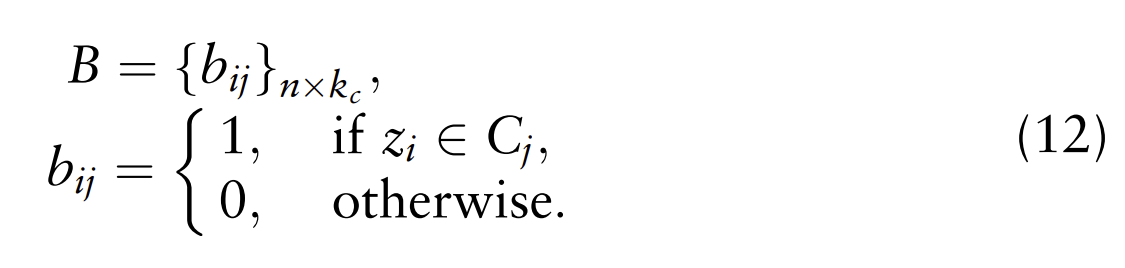
这表明,只有当一个节点是样本,另一个节点是包含该样本的聚类时,两个节点之间存在边。
二分图集成聚类
在二分图生成后,观察到这等同于在谱聚类中解决广义特征值问题(Shi和Malik,2000),可以表示为:
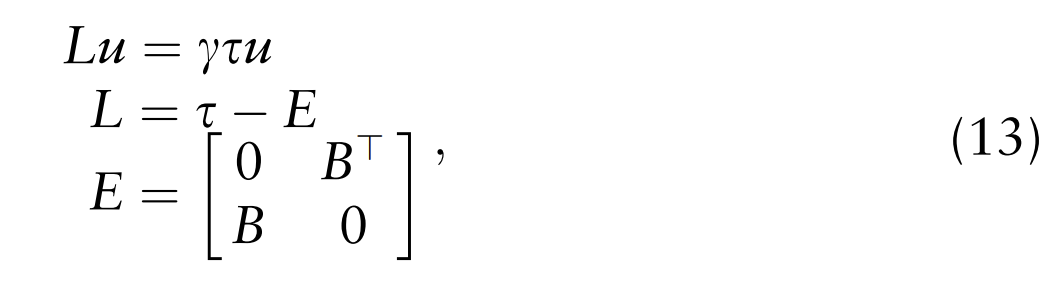
其中L是拉普拉斯矩阵,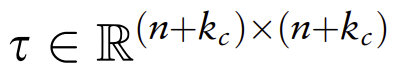 是度矩阵,E是G的完整亲和矩阵,n + kc是G中的节点数,因为Z和
是度矩阵,E是G的完整亲和矩阵,n + kc是G中的节点数,因为Z和![]() 是G中的节点。
是G中的节点。
然而,将G作为一般图形并不适用于大规模数据集的计算。已经证明,在图G上解决特征值问题等价于在一个更小的图上解决它(Li等,2012)。因此,为了降低复杂性以利用二分图结构,作者采用转移切割(Li等,2012)来通过将特征值问题与G转移到更小图GR(具有kc个节点)来有效地划分图G。特别地,GR定义为:

其中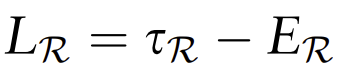 是GR的拉普拉斯矩阵。根据GR的前k个特征向量{v1, v2, ..., vk},可以计算G的前k个特征向量{u1, u2, ..., uk}(Huang等,2019)。最后,通过堆叠{u1, u2, ..., uk},对新矩阵执行K均值聚类,提供一致性聚类结果。
是GR的拉普拉斯矩阵。根据GR的前k个特征向量{v1, v2, ..., vk},可以计算G的前k个特征向量{u1, u2, ..., uk}(Huang等,2019)。最后,通过堆叠{u1, u2, ..., uk},对新矩阵执行K均值聚类,提供一致性聚类结果。
自编码器部分是深度的,可是学到潜在表示Z之后,构造二分图,利用二分图集成聚类,这一部分就不是深度了,不能优化回调。















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









