







SummarizedExperiment object
SummarizedExperiment for Coordinating Experimental Assays, Samples, and Regions of Interest
SummarizedExperiment 对象能够存储多个样本的观测值,同时存储用于描述观测值和样本的元数据(metadata)。SummarizedExperiment 能够紧密结合样本,观测值与对应的metadata,比如,如果在分析中删除了某个样本,则可同步删除与这个样本相关metadata。
SummarizedExperiment 对象
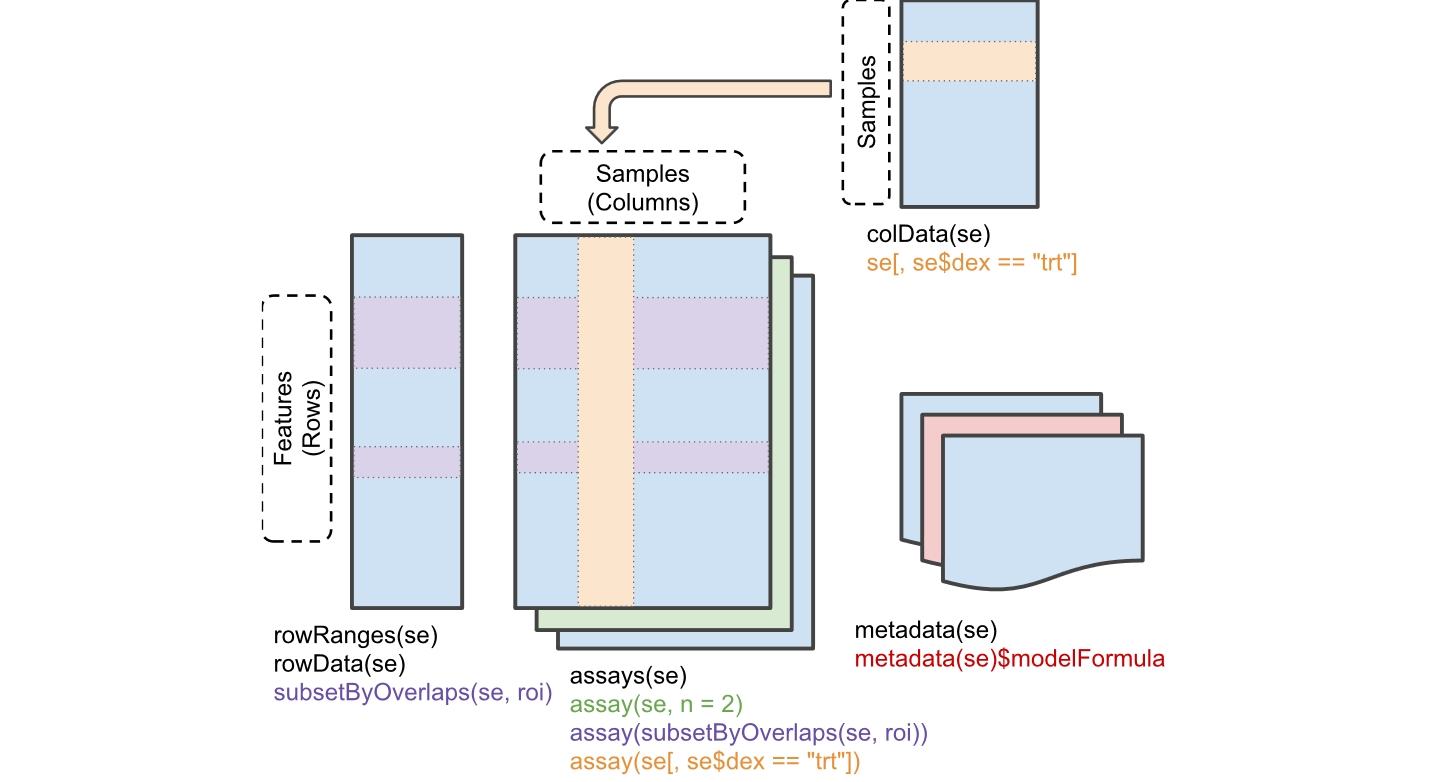
SummarizedExperiment 对象本身是一个 gene x sample matrix,这个 gene x sample matrix 储存在 assay中,同时在这个矩阵的基础上增加了对 gene 的注释信息 和 对 sample 的注释信息,也就是两个 metadata matrix。在这个gene x sample 矩阵中,行为 features name,例如:基因名、基因ID、SNP位点等,行的metadata为 DataFrame 对象,可以通过 rowData() 命令访问; 在这个DataFrame 对象中,行为基因名,列为基因的相关的注释信息,例如转录起始位点、终止位点、染色体信息等。列为 samples,列的metadata中包含对样本的一系列表型信息,比如年龄、大小、性别等,可以通过 colData() 访问。需要注意的是一个SummarizedExperiment 对象是允许同时包含多个 gene x sample matrix 的,也就是多个 assyas
SummarizedExperiment 对象还有一个子对象 - RangedSummarizedExperiment,两者的区别是,在 SummarizedExperiment 对象中,行的注释信息储存为 DataFrame 格式;而RangedSummarizedExperiment 中行的注释信息储存为 GRanges 格式,通过 rowRanges() 访问。
用 ArchR 对 scATAC 数据分析,通过 getMarkerFeatures() 命令会得到一个对每个 peaks 分析后的统计信息,以该数据为例进行如下操作:
1. 访问 SummarizedExperiment 对象
marker_peaks_group <- getMarkerFeatures(
ArchRProj = projHeme5,
useMatrix = "PeakMatrix",
groupBy = "predictedGroup",
bias = c("TSSEnrichment", "log10(nFrags)"),
testMethod = "wilcoxon"
)
# 1. SummarizedExperiment 对象,行为peaks,共49527个peaks;列为不同细胞类型,共27个
> marker_peaks_group
class: SummarizedExperiment
dim: 495247 27
metadata(2): MatchInfo Params
assays(7): Log2FC Mean ... AUC MeanBGD
rownames(495247): 1 2 ... 495246 495247
rowData names(4): seqnames idx start end
colnames(27): Adipocyte Chondrocyte 1 ... VSMC 1 VSMC 2
colData names(0)
# metadata: 分析使用的各种 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章
















 2552
2552











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









