







PAGA(Partition-based graph abstraction)轨迹分析功能包含在Scanpy软件中,是一种基于python的软件,对于没有python使用经验的人来说用起来还是比较吃力的。2019年发表的文献对其工作原理进行了详细阐述,下面是我在做轨迹分析时的一些理解。
在聚类分析中我们寻找的是细胞间的离散属性,也就是细胞之间的不同,而估计分析中我们寻找的是连续属性,也就是细胞之间的相同之处。PAGA是一种基于图形的分析方法,先通过 Louvain algorithm算法对细胞进行降维,生成低纬度的聚类图,基于聚类图进一步分析不同细胞类群之间的关系。下边是我用自己的数据生成的肌细胞谱系的PAGA轨迹分析结果,其实与其说是PAGA轨迹分析图,不如说是PAGA轨迹关系图。
1. 从seurat到PAGA
个人比较习惯R studio的操作环境,所以细胞质控、数据预处理、注释等全部是用Seurat软件分析的(当然scanpy也有同样的功能),将处理后的数据先转为example.h5Seurat,在转为example.5had文件,PAGA可直接分析.h5ad文件.
remotes::install_github("mojaveazure/seurat-disk") ##安装seuratdisk软件包
library(SeuratDisk)
SaveH5Seurat(example.cell, "example.cell.h5Seurat") ##example.cell直接转为h5Seurat格式保存
Convert("example.cell.h5Seurat", dest = "h5ad") ##h5Seurat转为h5ad
同样的
#####读取h5ad文件
##h5ad转为h5seurat
Convert('example.cell.h5ad', "h5seurat",overwrite = TRUE,assay = "RNA")
example.cell <- LoadH5Seurat("example.cell.h5seurat")2. PAGA图
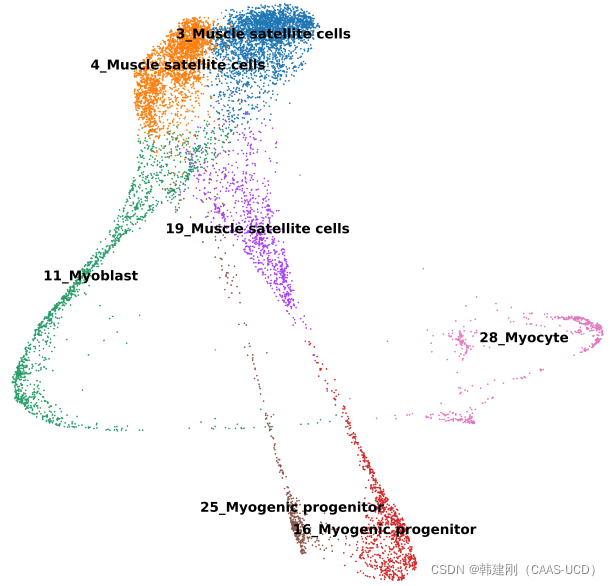
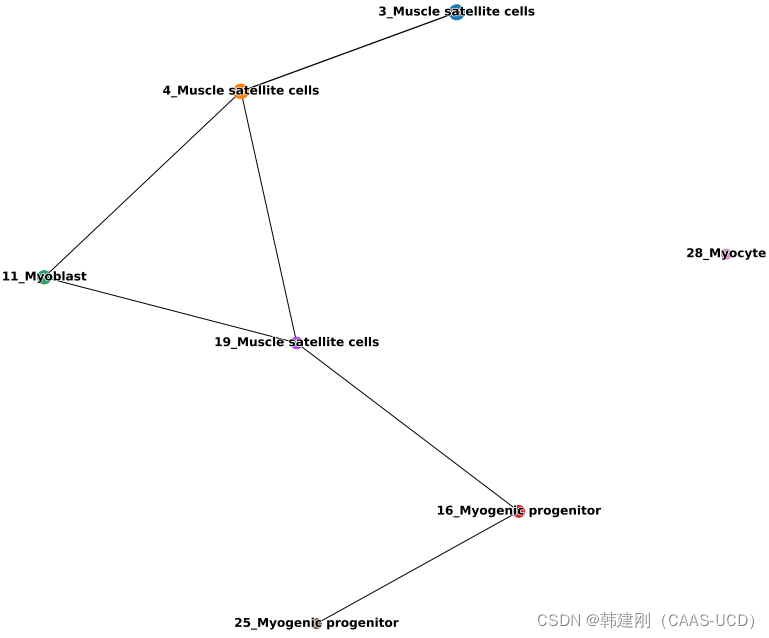
左图是PAGA软件对全部细胞的分群结果,右图是轨迹关系推断(n_neighbors=10;threshold=0.7),可以看到在两张图片中不同细胞类群的位置关系还是比较一致的。轨迹关系结果仅仅只是简单的点-线图,那么如何理解这张图?首先点代表一个细胞类群,两点之间的连线代表两个细胞类群之间有关系,线的长短反映两个细胞类群在聚类图上的位置关系(并没有太重要的作用),线的粗细表示得到的轨迹关系的置信度(可以理解为显著性),线越粗,置信度越高。
3. 不同参数
做任何分析都会有不同的参数,PAGA轨迹分析也不例外,主要的参数有
resolution, n_pcs,n_neighbors,threshold
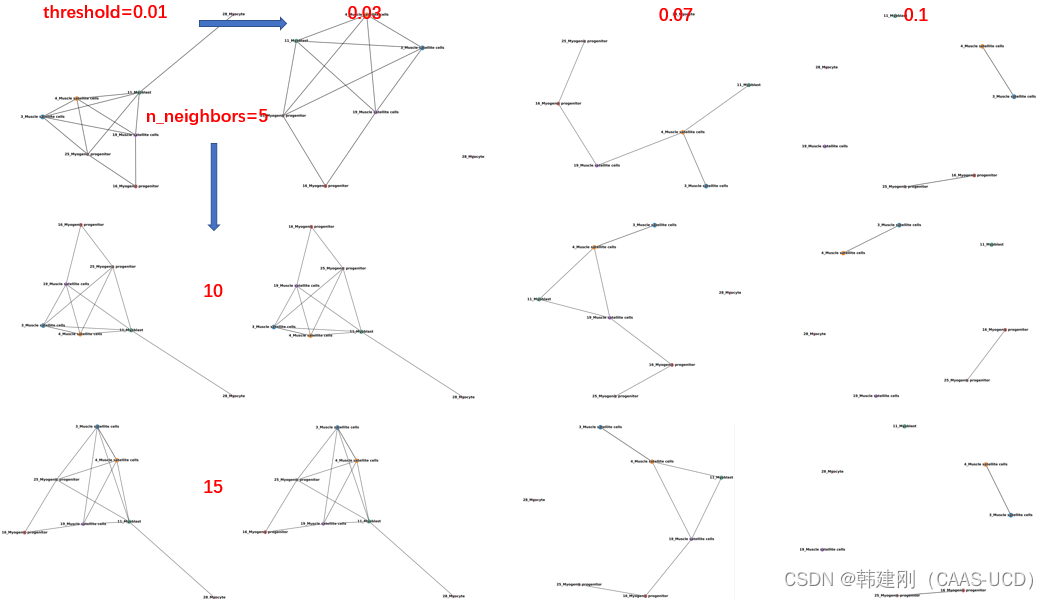
resulution, n_pcs两个参数与用seurat分析设置的参数感觉相似,可以设置为相同的值,应该不会对结果造成太大的影响。n_neighbors是计算参数,threshold是结果展示参数(个人理解,可能不太准确)。参数的选择是具有强烈主观性的,分析结果的好坏要看是否符合预期(主观性显著)。下面展示了两个参数不同的分析结果之间的比较。可以看到n_neighbors从5增加到15并不会对结果产生太大的影响。threshold分别设置成0.01,0.03,0.07和0.1,轨迹关系结构发生了显著的变化。从0.01到0.1,轨迹关系判别结果愈发严格,当我们对复杂组织、多种细胞类型的数据进行轨迹分析,可以调高threshold,而简单细胞群体的轨迹判别,可以设置成低的threshol。在这里选择n_neighbors=10,threshold=0.03的结果(主观)。
4. 几个有用的链接/教程
scanpy官网Trajectory inference for hematopoiesis in mouse — Scanpy documentation
两个教程


















 1115
1115

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









