







文章目录
IGV——基因组可视化(二)
本部分为基于生信修炼手册的《玩转基因组浏览器》系列的笔记大汇总,对于初级的 IGV 使用进行了更为高阶的补充。
常见的基因组浏览器包括 UCSC Genome Browser、Ensembl Genome Browser、JBrowse 和 IGV,其中前三者都是基于网页,而 IGV 不仅提供了 web 服务,还提供了独立的安装包,允许在本地运行,同时还提供了命令行版本的运行工具 igvtools。
IGV 支持芯片数据,NGS 数据,基因组注释等多种类型的数据,各种类型的信息以行为单位进行展示,称为 track,不同类型的数据可视化方式不同。
官网:https://software.broadinstitute.org/software/igv/
网页版:https://igv.org/app/
(一)参考基因组
1. 下载内置参考基因组
IGV 软件内置有多个物种的参考基因组,可以直接通过 IGV 工具下载载入。
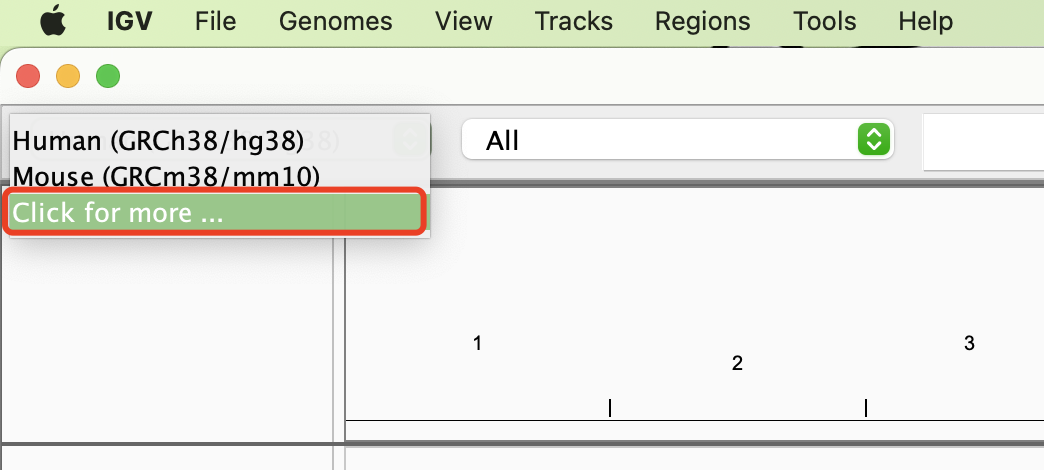
2. 自定义参考基因组
由于网络或其他原因,载入内置参考基因组时可能会失败,此时可以在本地构建参考基因组,使用时从本地导入即可,该方法即使在没有网络的情况下也可以使用。
构建本地参考基因组需要以下文件:
-
genome fasta:参考基因组的fasta文件,可以是一个文件包含了所有的染色体,也可以是一个目录,目录下每条染色体是一个单独的文件**(必须)**
-
cytoband file:染色体条带文件(可选)
-
gene annotation file:基因结构注释文件,支持bed、gtf、genePred 3种格式(可选)
-
alias file:别名,当fasta文件和基因结构中的染色体名称不同时,可以通过这个文件来进行映射(可选)
通常情况下,只需要基因组序列和基因结构文件即可满足需求。
注意:从版本 2.11.0 开始,通过“.genome” 格式指定参考基因组已被弃用,现在指定参考基因组为 JSON 文件(https://github.com/igvteam/igv/wiki/JSON-Genome-Format)。
(1)对于 fasta 文件,需要先建立后缀为 .fai 的索引。
# conda install -c bioconda samtools
samtools faidx hg19.fa
(2)根据文件 url 或路径创建基因组 JSON 文件。
必填字段为 “id”、“name”、“fastaURL” 和 “indexURL”,所有其他字段都是可选的。
URL 属性(所有以 url 结尾的字段)可以是绝对或相对文件路径,相对路径被解释为相对于基因组 json 文件的位置,如以下定义假定注释文件 gencode.v24.genes.gtf.gz 与 json 文件位于同一目录。
{
"id": "hg19",
"name": "Human (CRCh37/hg19)",
"fastaURL": "https://s3.amazonaws.com/igv.broadinstitute.org/genomes/seq/hg19/hg19.fasta",
"indexURL": "https://s3.amazonaws.com/igv.broadinstitute.org/genomes/seq/hg19/hg19.fasta.fai",
"cytobandURL": "https://s3.amazonaws.com/igv.broadinstitute.org/genomes/seq/hg19/cytoBand.txt",
"aliasURL": "https://s3.amazonaws.com/igv.org.genomes/hg19/hg19_alias.tab",
"tracks": [
{
"name": "Gencode v24 genes",
"url": "gencode.v24.genes.gtf.gz"
},
]
}
通过创建本地化的参考基因组,可以避免网络差用不了IGV的情况,也大大扩展了IGV支持的物种列表。
(3)载入自制参考基因组
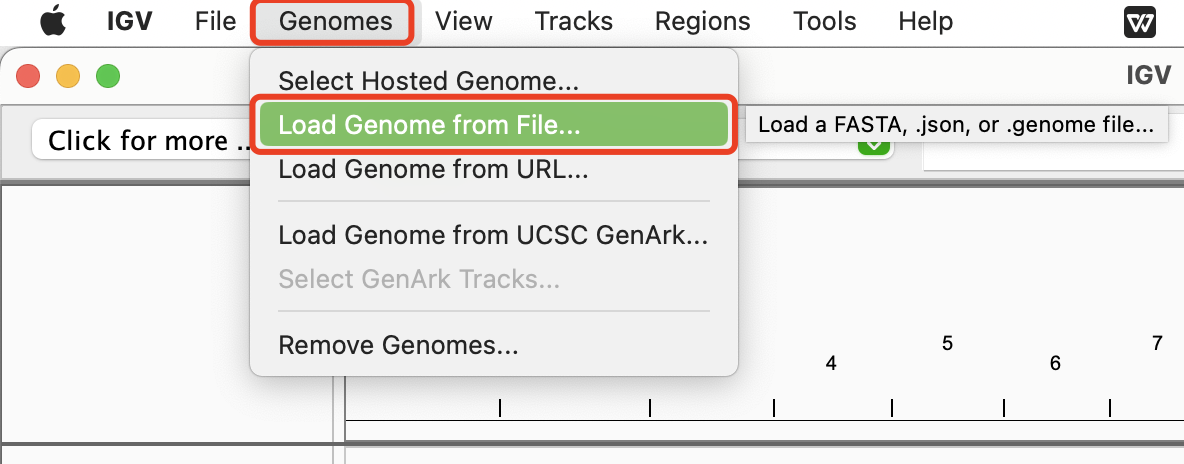
(二)基因结构
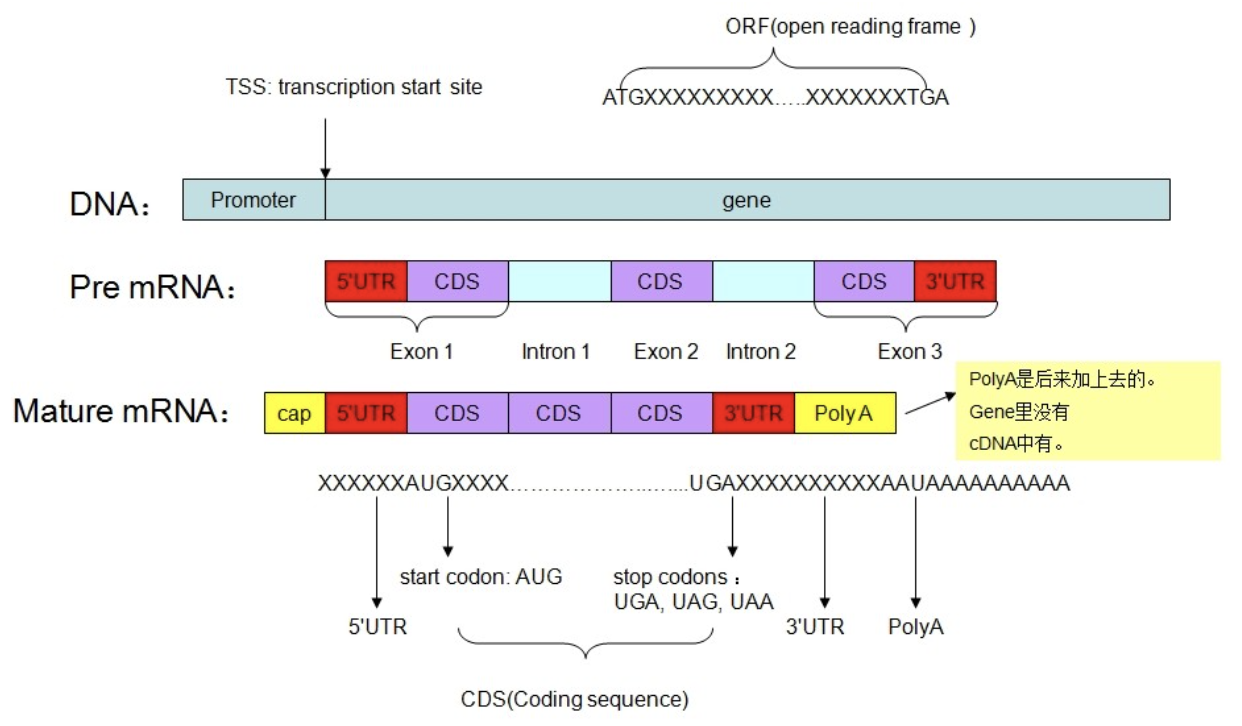
基因结构是最基本的基因组注释信息,GFF、GTF、BED 文件通常用于存储基因结构信息,其本质为染色体坐标。
IGV 要求导入数据必须为排序后的结果,同时还需要对排序后的文件建立索引,使检索速度更快,排序后的 gtf 文件需要与其索引放置在同一目录下。
sort -k1,1 -k4,4n -k5,5n hg19.gtf > hg19.sort.gtf
igvtools index hg19.sort.gtf
导入 gtf 文件后,所有转录本会折叠在同一行展示,下方为对应的 gene name,该展示方式为 Collapsed ,节省空间,但难以区分不同转录本。
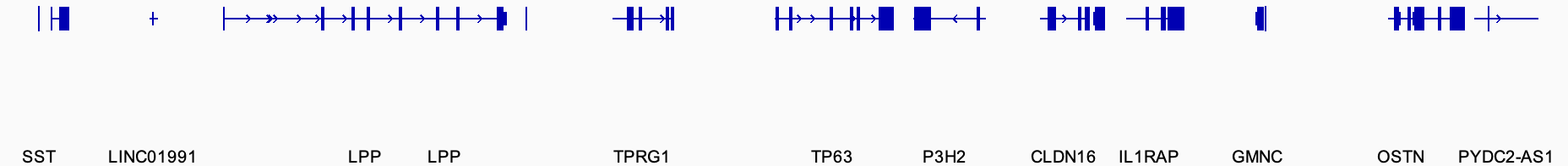
可以通过右键该 track 选择不同的展示方式。
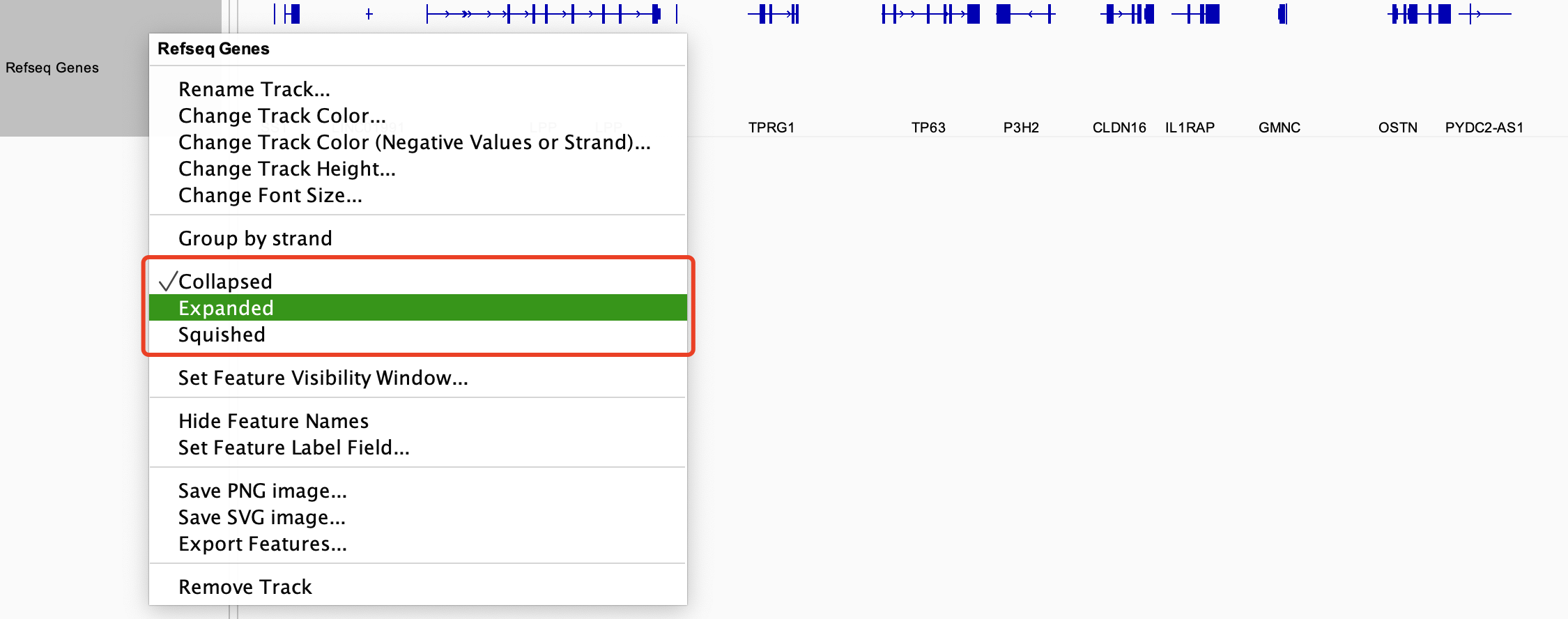
其中,Expanded 模式下转录本区分的最清楚,但是占据的空间很大,Squished 则是一种折中方案,抛弃了gene_name,进一步压缩了空间。
Expanded 模式:
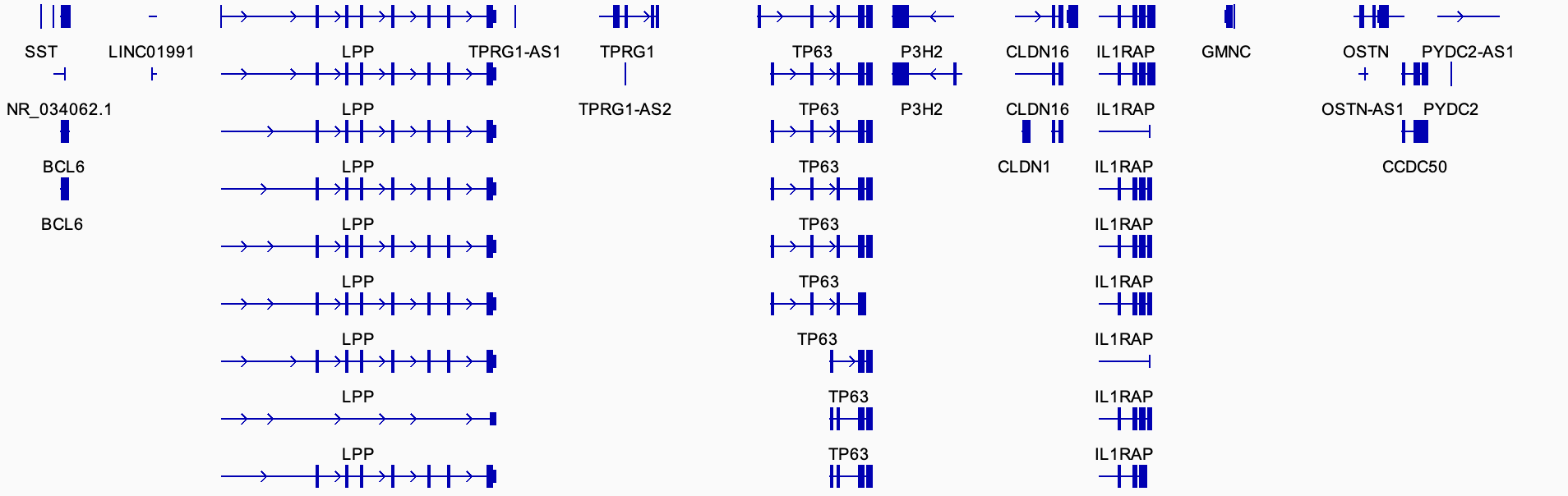
Squished 模式:
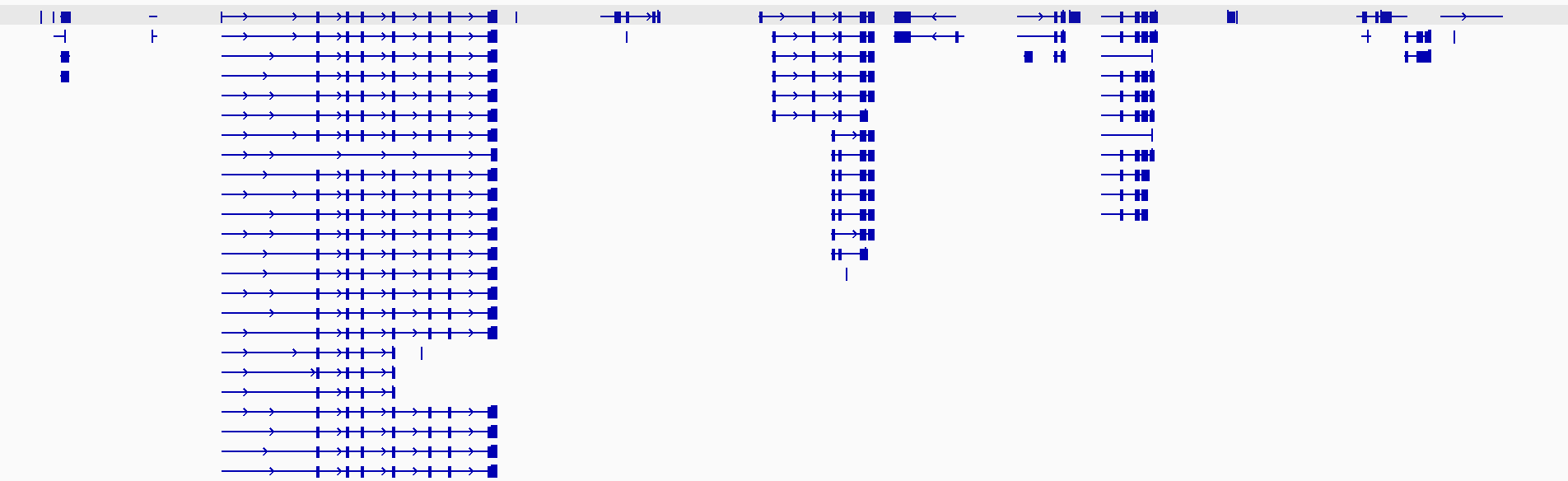
其中,每一条转录本,由3种元素构成:

-
矩形:表示exon区域,较窄的矩形区域表示的是UTR区域,对于蛋白编码RNA,当GTF文件中提供了UTR或者CDS的区间时,会自动计算出UTR区域并进行标注
-
线条
-
箭头:表示基因的正负链信息,向右的箭头表示正链,向左的箭头表示负链
(三)BAM 文件
bam 文件记录了 reads 比对到参考基因组的详细信息,使用 IGV 可视化不仅可以灵活的选择想要查看的基因组区域,还能够查看测序深度的分布,exon 的连接方式等更多的信息,bam 文件需要排序并建立索引之后才可以导入 IGV。
对于 bam 文件,IGV提供了以下3种视图:
- alignment track,展示序列的比对情况,默认情况下,与参考基因组精确匹配的碱基用灰色表示,错配的碱基则根据A,T,C,G映射到不同的颜色。在reads中,进一步对插入和缺失进行了标记,插入的碱基用
Ⅰ表示,缺失用-表示。
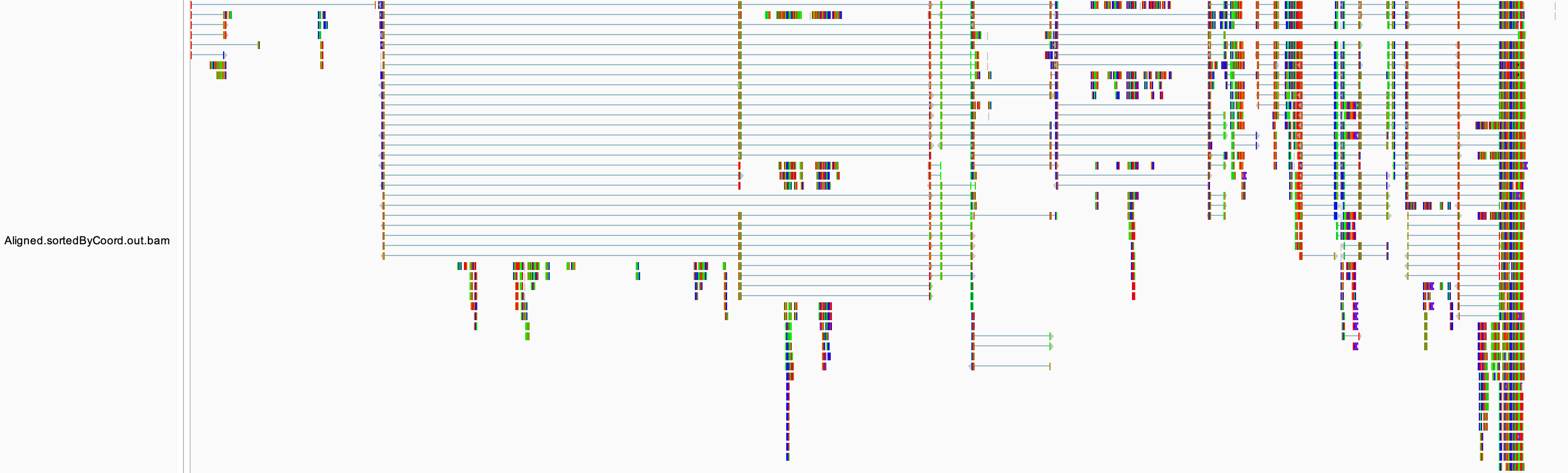
- coverage track,柱状图展示每个位点的测序深度信息。
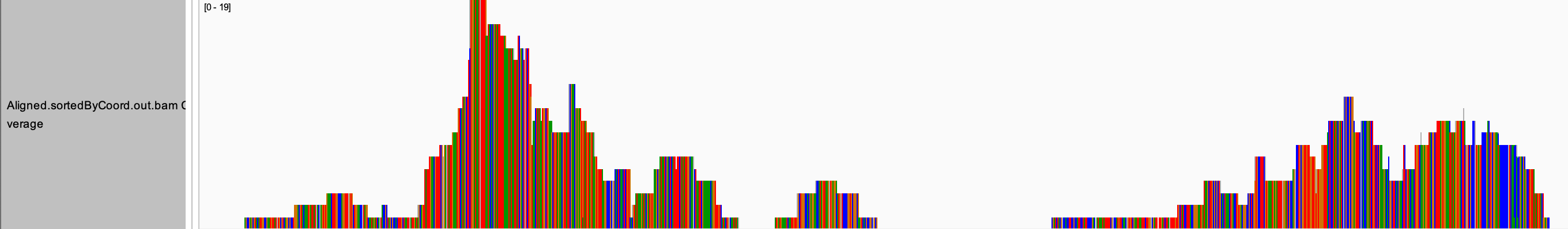
- splice junction track,对于RNA_seq的数据,由于内含子的存在,reads比对到基因组上需要跳过内含子,splice junction track 用曲线将外显子边界连接起来。
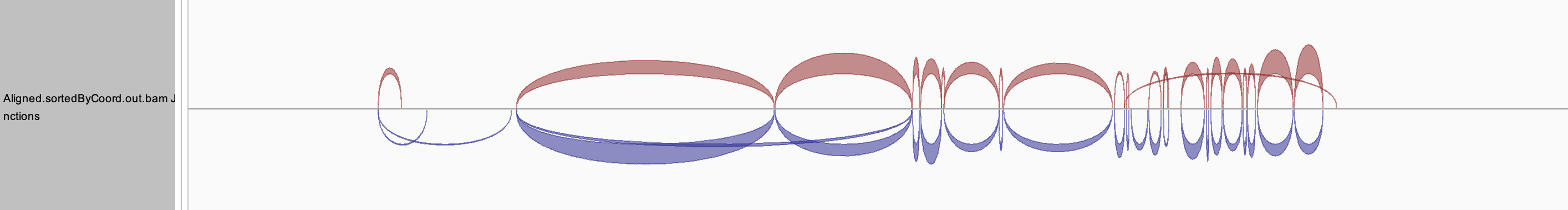
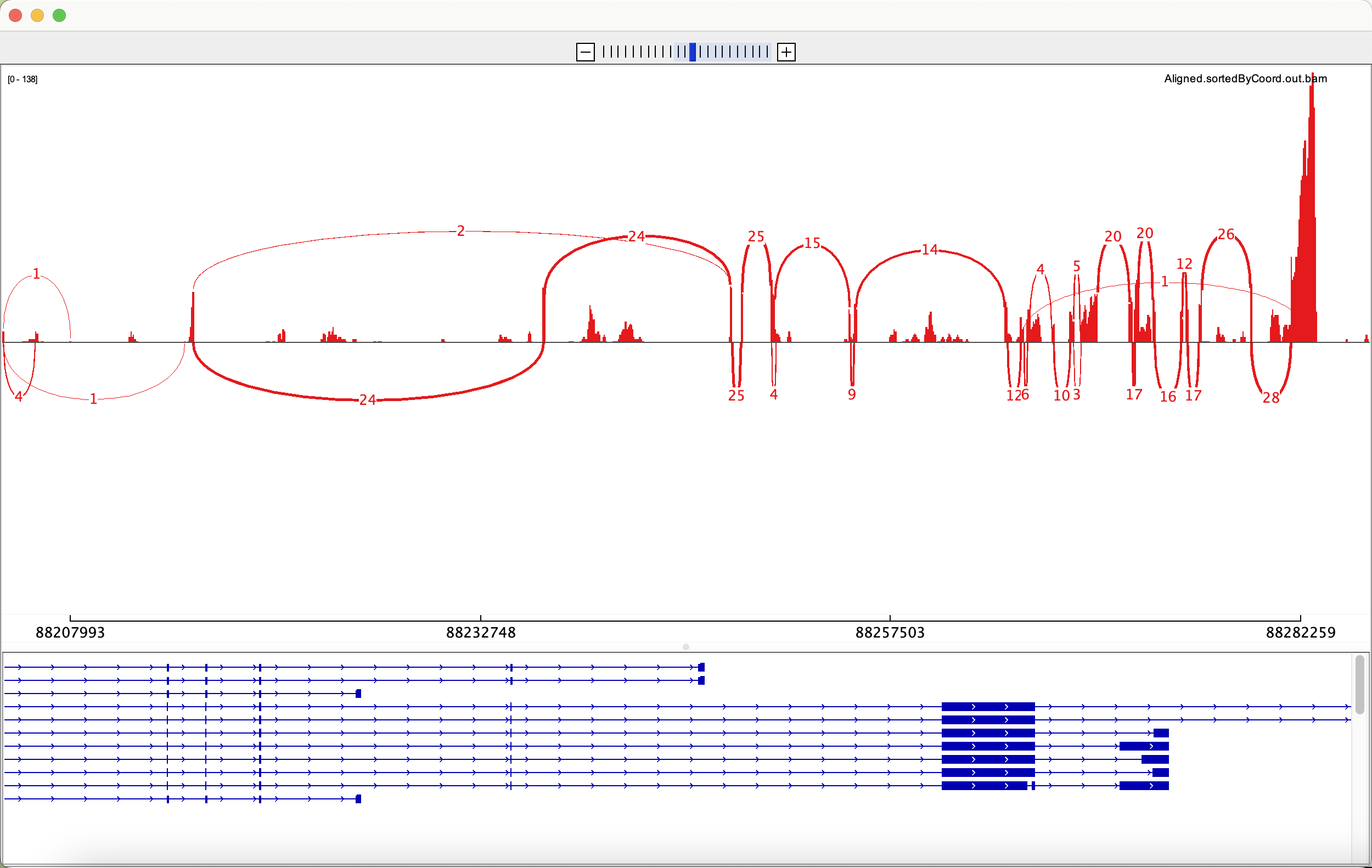
同时,还可以通过右键 -> Sashimi Plot 来实现可变剪切可视化。
(四)BED 文件
bed 文件用于记录染色体区域信息,其可以具有多个变种,如转录本结构可以用 bed12 格式来记录,peak calling 的结果可以用 bed6+4 来记录,常见的 bed 相关格式有 bed、narrowpeak、broadpeak 和 bigbed,均可以导入 IGV 进行查看。
其中,narrowpeak 和 broadpeak 常用于存储 peak calling 的结果,分别为 bed6+4 和 bed6+3 格式,即分别为 10 列和 9 列,bigbed 是二进制的 bed 文件。
由于 IGV 要求导入数据必须为排序后的结果,此处需要对 bed 文件进行排序,同时还需要构建索引,可以直接按照 IGV 的提示完成索引构建。
sort -k1,1 -k2,2n input.bed > sort.bed
将 bed 文件与其索引置于同一路径下,并将 bed 文件导入 IGV 中。
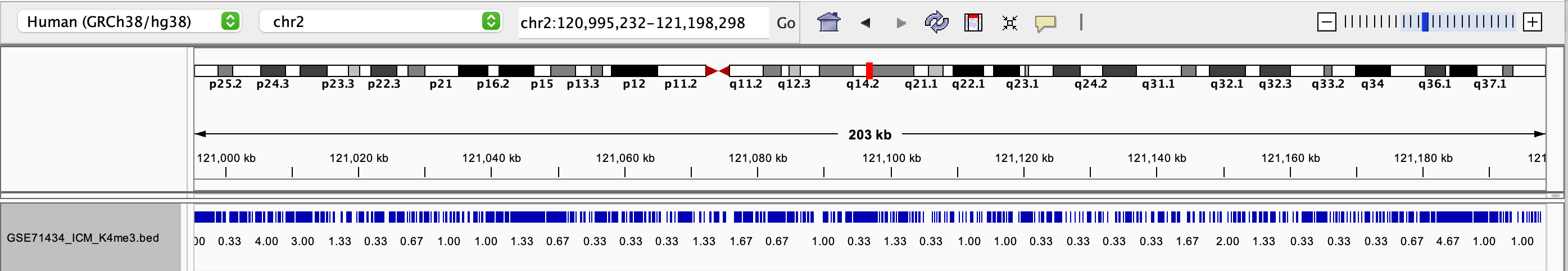
bed 文件中的每一行为一个染色体区域,对应图中蓝色的矩形区域。如果 bed 文件的第四列提供了染色体区域名称,在矩形区域的下方会显示对应的标识符,可以根据标识符进行检索。
bed 文件可以用于快速查找感兴趣的基因组区域,最经典的应用场景为将 peak calling 和对应样本的文件同时导入 IGV 即可快速查看 peak 区域的测序深度分布情况。
补充:
- bigbed 与 bed 格式可以通过 UCSC 提供的工具来进行格式转换:
# conda install -c bioconda ucsc-bedtobigbed
# conda install -c bioconda ucsc-bigbedtobed
# bed to bigbed
sort -k1,1 -k2,2n input.bed > sort.bed
bedToBigBed sort.bed hg19.chrom.sizes out.bigbed
# bigbed to bed
bigBedToBed input.bigBed out.bed
- chrom.sizes 文件可以直接在 igvtools 的安装目录下的
lib/genomes文件夹中找到,也可以通过如下命令生成染色体长度文件 chrom.sizes:
# conda install -c bioconda ucsc-twobitinfo
# conda install -c bioconda ucsc-fatotwobit
faToTwoBit hg19.fa hg19.2bit
twoBitInfo hg19.2bit stdout | sort -k1,1 -k2,2n > hg19.chrom.sizes
(五)TDF 文件
将 bam 文件导入 IGV 之后,可以直观的查看测序深度的分布情况,但是直接导入 bam 文件会占用较大的内存,如果只是想要查看测序深度信息,可以将其转化为 tdf 文件格式来进行查看。
相比 bam 文件,tdf 文件更小,且导入和查看也更快速,tdf 文件是一种类似 bedgraph 格式的二进制文件,用窗口的方式来记录测序深度信息,可以通过 igvtools 来生成 tdf 文件。
# tdf 默认以 25bp 为窗口,统计该窗口内测序深度的平均值
igvtools count input.bam out.tdf hg19.chrom.sizes
# 修改窗口大小,窗口越大,对应的文件大小越小,分辨率也低
igvtools count -w 50 input.bam out.tdf hg19.chrom.sizes
其中,第一个参数为输入文件,支持bam,sam等格式;第二个参数为输出文件,支持 tdf 和 wig 两种格式;第三个参数为基因组的 ID 或者保存染色体大小的 chrom.sizes 文件。
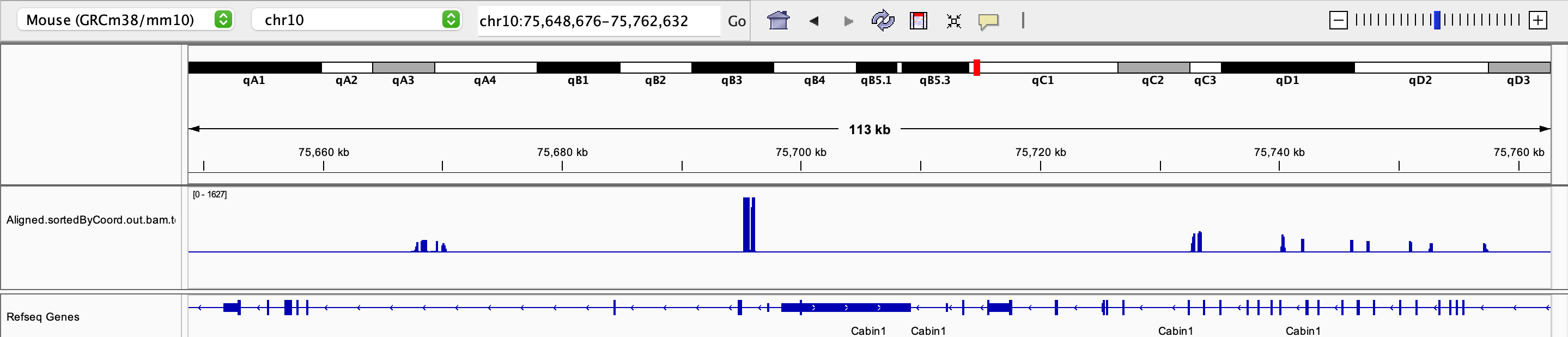
补充:
- bedGraph vs TDF vs bigwig
在统计区域上,bigwig > TDF > bedGraph,均以窗口的方式来统计测序深度(bam 以单个碱基为单位),三种文件在小分辨率下几乎没差异,在大分辨率下就会看出细节差异。
小分辨率:三者峰型基本一致。

大分辨率:

整体上统计区间:
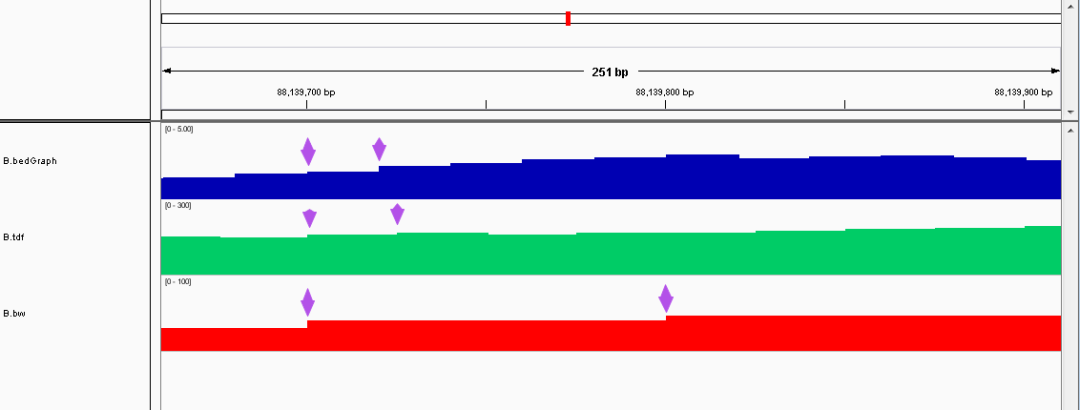
在文件大小上,bam > bedGraph > TDF > bigwig。

- 将 tdf 文件转换为 bedgraph 纯文本格式
igvtools tdftobedgraph input.tdf out.bedgraph
(六)序列比对
IGV 通过调用 UCSC 的 Blat 软件(https://genome.ucsc.edu/cgi-bin/hgBlat?command=start)来实现序列比对,在IGV中,通过工具栏的 Tools -> BLAT 菜单,可以自定义输入查询序列,直接在该输入框中粘贴查询序列的碱基即可。
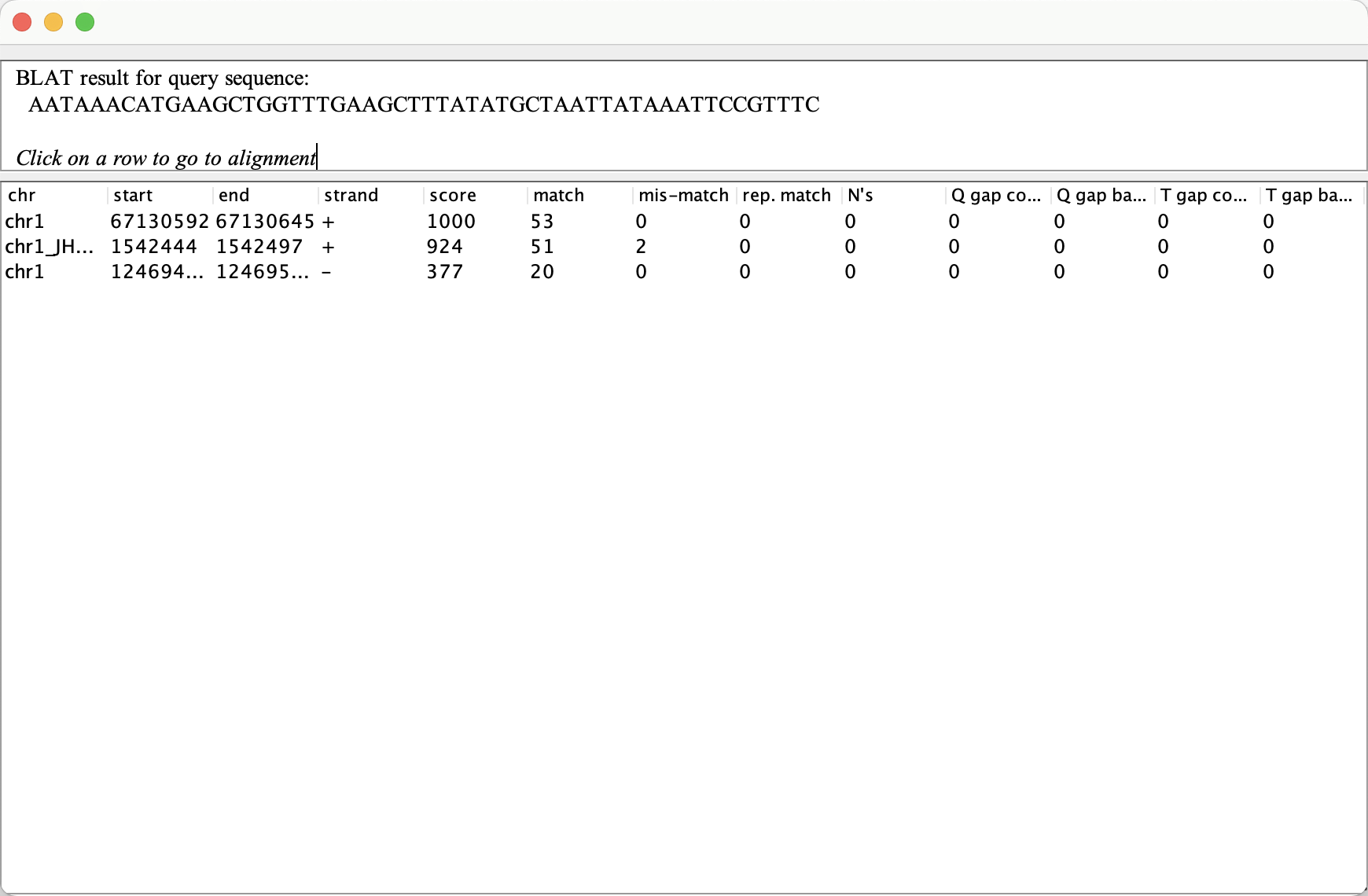
鼠标左键选中每一行,可以在基因组浏览器中展示比对结果,其中 Blat 的 track 则显示的是查询序列的比对位置。
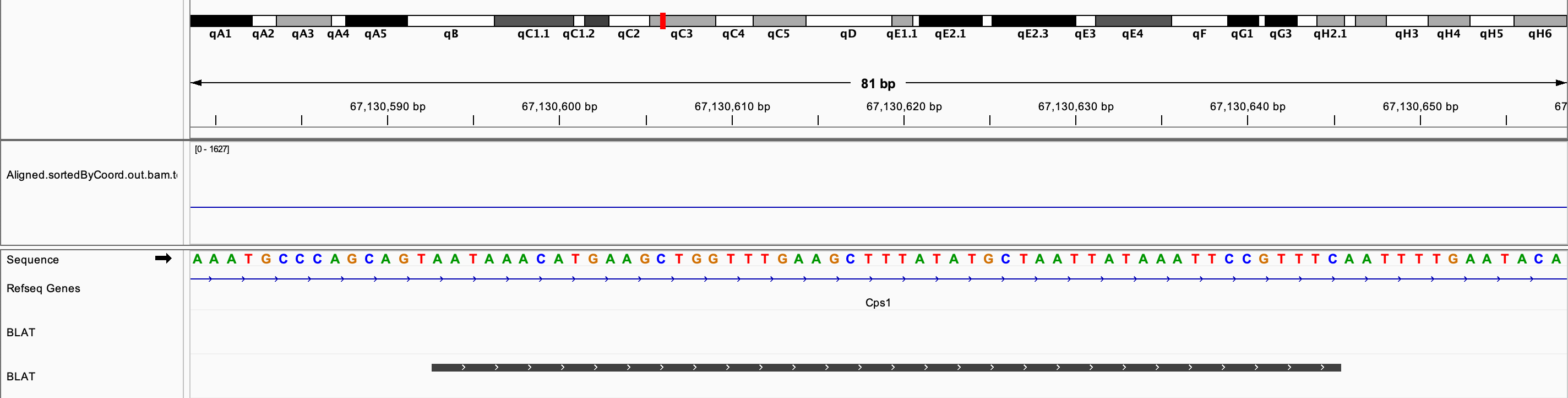
除了上述用法外,IGV 还可以对 bam 文件中的 reads 进行比对,对基因结构中的某个特征 exon、intron 进行比对等等,具体可参考 http://software.broadinstitute.org/software/igv/BLAT。
(七)motif
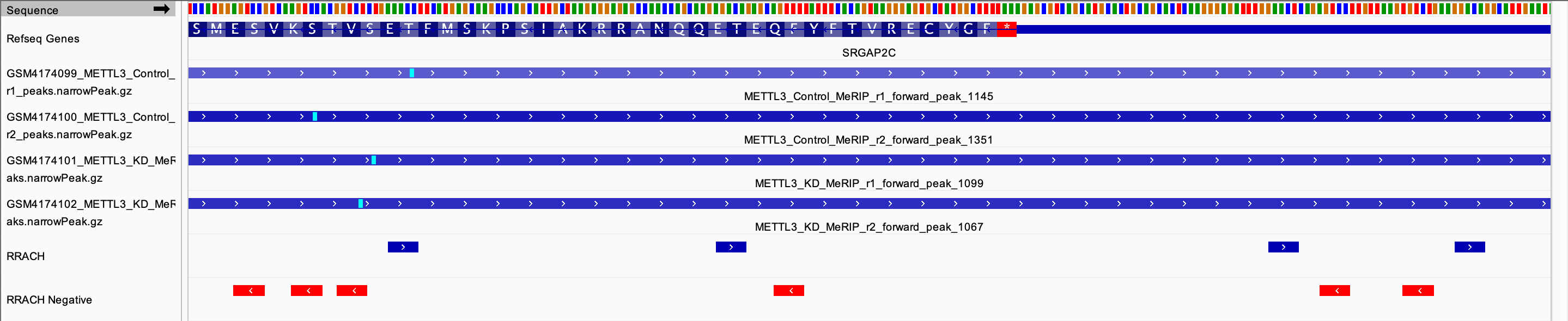
本部分以 RNA 甲基化测序 m6A_seq 为例展示 IGV 的 motif 结合位点查找功能,已知 m6A 修饰位点的 motif 序列为 RRACH,通过 peak calling 可以识别到包含 m6A 修饰位点的 peak 区域,此时需要预测在 peak 区域中发生了甲基化修饰的 A 碱基,可以通过 IGV 的 motif 查找功能确定候选位点。
示例数据来源:GSE140557,格式为 narrowpeak,保存的是 peak calling 的结果。
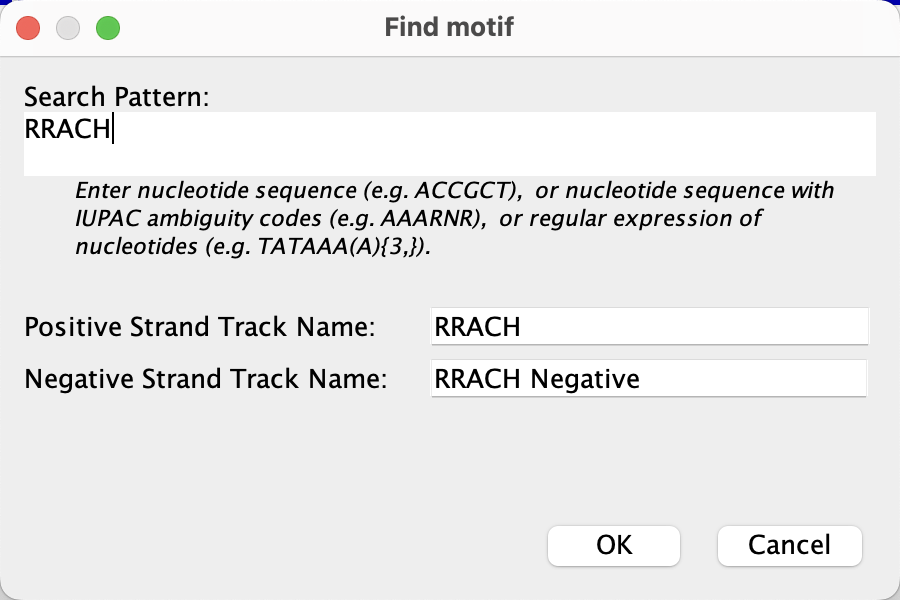
在IGV中,通过工具栏的 Tools -> Find Motif 功能在基因组的正负链分别查找潜在的motif结合位点。
结果如下:
补充 —— igvtools
在使用 conda install -c bioconda igvtools 安装 igvtools 时,直接在终端运行 igvtools 可能会出现报错,可能原因与 Java 版本不兼容。

此时可以通过 IGV 工具的图形界面 直接完成 igvtools 的运行,而无需使用命令行。
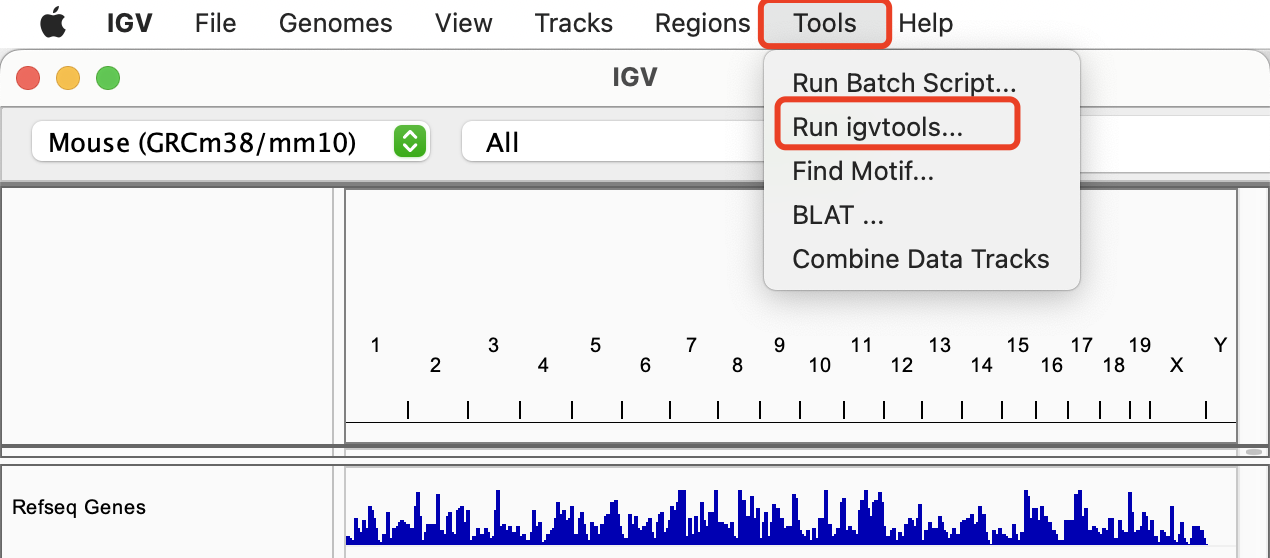
通过修改对应的 Command 选项即可以完成 igvtools 的不同分析。

参考
- 初识IGV:https://cloud.tencent.com/developer/article/1624537
- 自定义IGV的参考基因组:https://cloud.tencent.com/developer/article/1624542
- 使用IGV查看基因结构信息:https://cloud.tencent.com/developer/article/1624540
- IGV展示bam文件:https://cloud.tencent.com/developer/article/1624543
- 展示bed文件:https://cloud.tencent.com/developer/article/1624547
- tdf文件:https://cloud.tencent.com/developer/article/1624545
- IGV进行序列比对:https://cloud.tencent.com/developer/article/1624557
- 利用IGV查找motif结合位点:https://cloud.tencent.com/developer/article/1624559
进阶教程
除了基因结构,测序深度的可视化外,IGV 也可以展示基因组变异信息:
-
查看MAF文件:https://cloud.tencent.com/developer/article/1624549
-
VCF文件的可视化:https://cloud.tencent.com/developer/article/1624548
IGV 可以以热图的形式展示 CNV 分布和基因表达量的数据:
-
查看CNV分析结果:https://cloud.tencent.com/developer/article/1624553
-
展示RNA_seq中的基因表达量:https://cloud.tencent.com/developer/article/1624554
IGV 支持动态查看 gwas 分析结果,以曼哈顿图的形式展示所有位点的关联分析结果:
- 查看gwas结果:https://cloud.tencent.com/developer/article/1624551
除了展示 GTF,bed 等常规格式的基因结构信息,IGV 还可以展示 RNA 的二级结构:
- 展示RNA二级结构:https://cloud.tencent.com/developer/article/1624555
如果对于多个基因组区域感兴趣,可以使用 IGV 的分面功能来实现同时查看:
- 分面操作:https://cloud.tencent.com/developer/article/1624546

















 2765
2765

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









