







写在前面
我们《复现SCI文章系列教程》专栏现在是免费开放,推出这个专栏差不多半年的时间,但是由于个人的精力和时间有限,只更新了一部分。后续的更新太慢了。因此,最终考虑后还是免费开放吧,反正不是什么那么神秘的东西。原本就是一个套路的文章,此外,这篇文章也相对比较简单。在此章节以前,还有一个WGCNA的分析,你若需要可以看**WGCNA分析 | 全流程分析代码**
目前全部开放链接:
原付费:复现SCI文章系列教程文章
- 订阅《复现SCI文章系列教程》须知
- 复现SCI文章系列 | 第一篇文章复现:1. 文章讨论与文章分析套路讲解
- 2.1 材料与方法 (IF 7.3)
- 2.2 数据集下载 (IF 7.3)
- 2.3 数据去重和标准化(附送去批次效应)
- 2.4 差异分析
- 2.5 加权基因共表达分析(WGCNA)
- 2.6 PPI网络分析
本期推文内容
2.7.1 章节总结
在前的教程中,我们已经获得差异基因(2.4 差异分析)和获得与纤维化相关的模块基因。此教程,我们做功能富集分析。但是,此数据问题依旧是很大的影响因素,严重影响后续的分析。
2.7.2 文章结果内容
- GO和KEGG富集分析结果

- 分析结果图
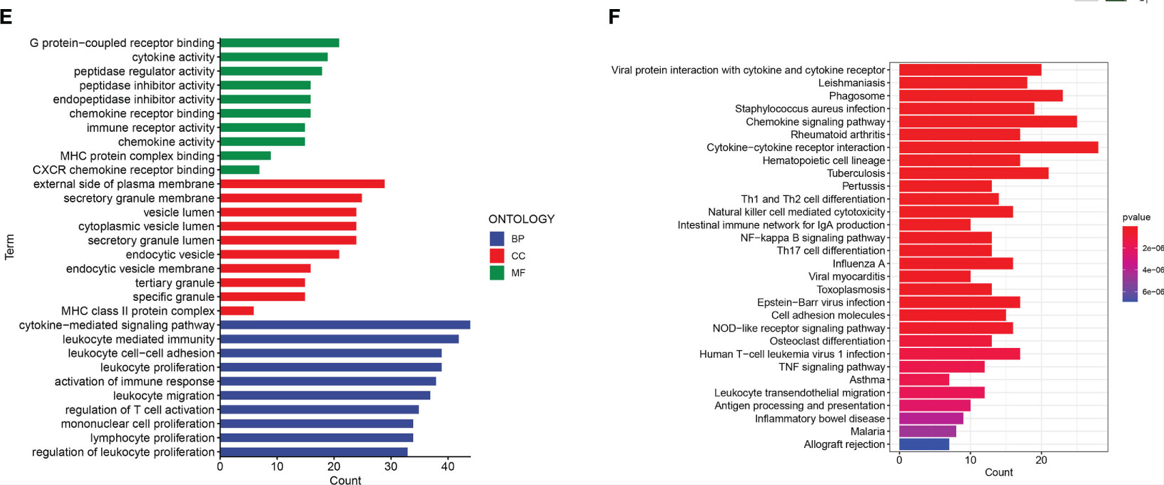
2.7.3 取交集
根据文章分析流程,将DEGs和WGNCA分析获得的结果去交集,获得的交集基因进行后续分析。

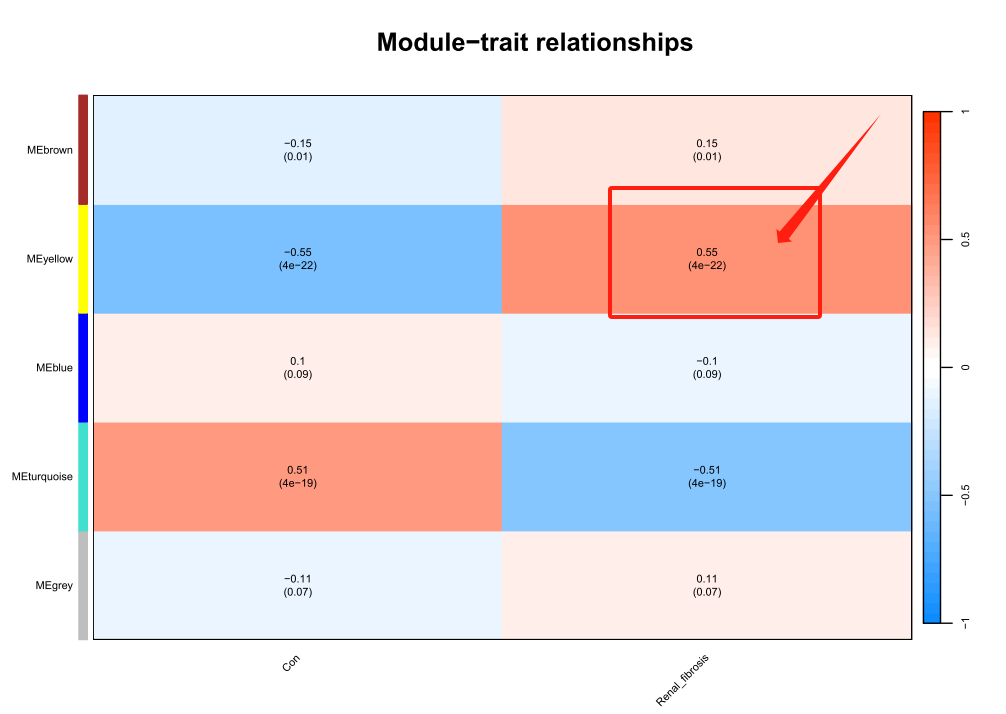
在差异分析中,我们获得600多个DEGs,在WGCNA分析中,与纤维化相关的模块为“yellow”。
共有200多个基因。

WGCNA分析全部结果。
2.7.4 获得交集并绘制韦恩图
汇总数据
绘制vennplot,可以是哟TBtools绘制韦恩图,很是方便。或是直接使用我们前提教程进行绘制,基于R语言绘制VennPlot图 | 可以绘制大于等于7个类别的码.
根据筛选出来的关键模块,采用GS>0.2,MM>0.8筛选高度关联基因并与差异基因取交集。我们在自己做分析时,或在写论文时,其实这些参数可以写进论文中,对读者是比较友好的。
但是这里的结果真的是出乎我的意料。我们继续往下看。
绘制维恩图代码
setwd("E:\\小杜的生信筆記\\2023-复现期刊文章系列教程\\复现文章一分析\\05.功能富集分析")
library(VennDiagram)
library(ggplot2)
library(venn)
library(RColorBrewer)
readFlie=function(input,type,row=T,header=T){
# input 为读入文件的路径,type为读入文件的类型,格式为‘.txt’或‘.csv’,row=T,将文件的第一列设置为列名
library(data.table,quietly = TRUE)
if(type=='txt'){
dat = fread(input,header = header,sep='\t',stringsAsFactors = F,check.names = F)
if(row){
dat = as.data.frame(dat,stringsAsFactors = F)
rownames(dat) = dat[,1]
dat = dat[,-1]
}else{
dat = as.data.frame(dat,stringsAsFactors = F)
}
}else{
dat = fread(input,header = header,sep=',',stringsAsFactors = F,check.names = F)
if(row){
dat = as.data.frame(dat,stringsAsFactors = F)
rownames(dat) = dat[,1]
dat = dat[,-1]
}else{
dat = as.data.frame(dat,stringsAsFactors = F)
}
}
return(dat)
}
## 绘制venn图
wn_venn=function(list,col='black'){
# 定义颜色体系
library(RColorBrewer,quietly = TRUE)
#corlor = brewer.pal(8,'Dark2')
corlor = brewer.pal(8,"Accent")
# 绘制Venn图
library(VennDiagram, quietly=TRUE)
library(venn,quietly = TRUE)
if(length(list)<=11){
if(length(list)<=4){
graphics=venn.diagram(list,filename=NULL,fill = corlor[1:length(list)],
col = col,alpha = 0.5, cat.cex = 1.5,rotation.degree = 0)
grid.draw(graphics)
}else if(length(list)==5){
graphics=venn(list, zcolor = corlor[1:length(list)],box=F,ellipse =TRUE,cexil = 1, cexsn = 1)
}else{
graphics=venn(list, zcolor = corlor[1:length(list)],box=F,cexil = 1, cexsn = 1)
}
return(graphics)
}else{
print('The function only supports data of dimension 7 and below.')
}
}
## 保存图片,只支持ggplot对象
savePlots=function(path,plot,type=c('pdf','png','tiff')[1],width=10,height=8,dpi=300){
# path表示保存图片路径,需要加上相应的文件扩展名称
library(ggplot2)
if(type=='pdf'){
ggsave(filename = path,plot = plot,width = width,height = height,device = 'pdf')
}else if(type=='png'){
ggsave(filename = path,plot = plot,width = width,height = height,device = 'png',dpi = dpi)
}else{
ggsave(filename = path,plot = plot,width = width,height = height,device = 'tiff',dpi = dpi)
}
}
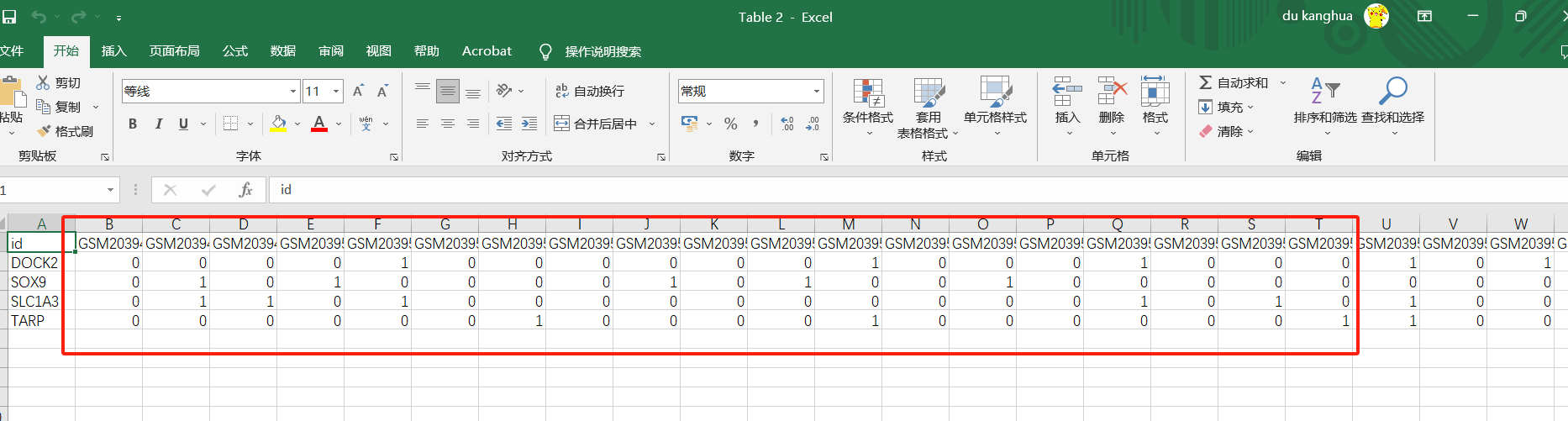
df <- readFlie("03_DEG_WGCNA交集数据.txt", type = "txt", row = F)
head(df)
df_list = list('DEGs'=sample(df$DEGs),'WGCNA'=sample(df$WGCNA))
# 绘制venn图
## 4维veen图
venn_2 = wn_venn(df_list[1:2])
# 保存图片
savePlots(path = 'DEGs_WGCNA_VennPlot.jpg',plot = venn_2,type = 'jpg',width = 8,height = 8,dpi = 300)
结果
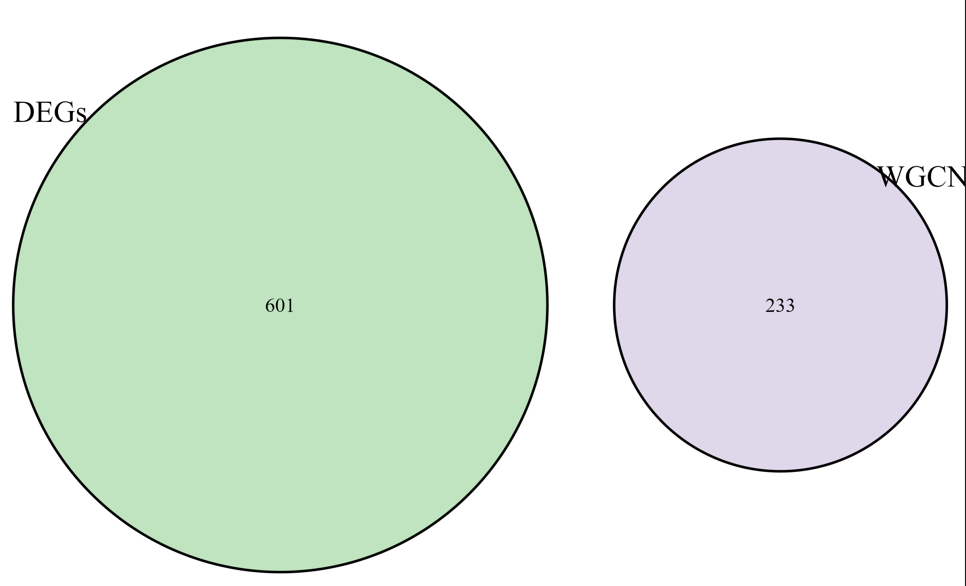
所以基因无任何交集。这个是什么原因呢???
现在我们又重新回去做分析。
WGNCA重新分析代码
寻找方案一
我们开始做WGCNA分析时,有过滤数值。
- 使不过滤的数据进行WGCNA分析。
寻找方案二
WGCNA分析,不去离散样本,所有样本进行分析。
我们这里不在坐重复演练,大体的流程就是这样。我们只需要学会这个思路就可以,不需要和文章一模一样的结果。
正常结果应该是这样的,一定会有交集。我们的只需要调整相关的参数即可。但是更具前面的分析就是离谱。
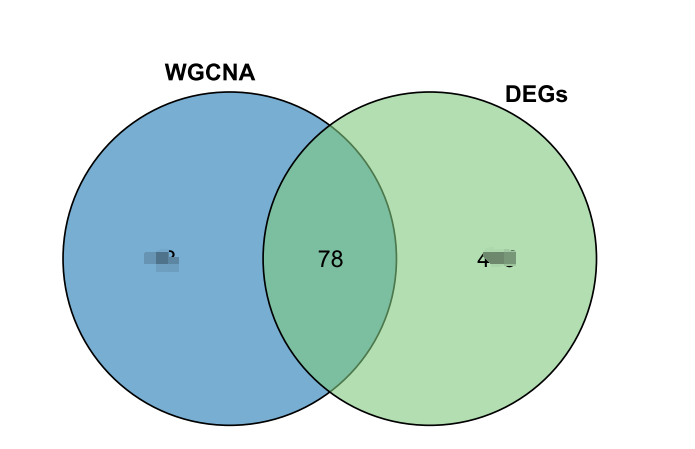
猜测
- 数据问题
我有仔细回去看了原文作者使用的数据,这是count值吗?但是这个数据怎么能这么小?以及与我们获得表达量直接是不对等,若是原文作者将FPKM值转换成count值,但依旧是不对等。
- 数据标准化或归一化的原因
自己认为,我们原始获得数据,已经是进行标准化后的数据了,应该是作者上传时就已经处理过。最大值在13…,但是原文作者依旧是进行标准化处理,那么我们也进行处理吧。

- 不知道,能力有限的原因??????
GO富集分析
setwd("E:\\小杜的生信筆記\\2023-复现期刊文章系列教程\\复现文章一分析\\05.功能富集分析")
library(stringr)
library(ggplot2)
library(clusterProfiler)
library(org.Hs.eg.db)
diffSig <- read.table("关键基因ID.txt",header=F,sep="\t",row.names=1)
DEG.gene_symbol = as.character(rownames(diffSig))
DEG.entrez_id = mapIds(x = org.Hs.eg.db,keys = DEG.gene_symbol,
keytype = "SYMBOL",column = "ENTREZID")
gene = bitr(DEG.gene_symbol, fromType="SYMBOL", toType="ENTREZID", OrgDb="org.Hs.eg.db")
DEG.entrez_id = na.omit(DEG.entrez_id)
DEG.entrez_id
write.csv(gene, "01_78.gene.SYMBOL_WNTREZID.csv")
#write.table(DEG.entrez_id,"DEG.entrez_id.txt",sep="\t")
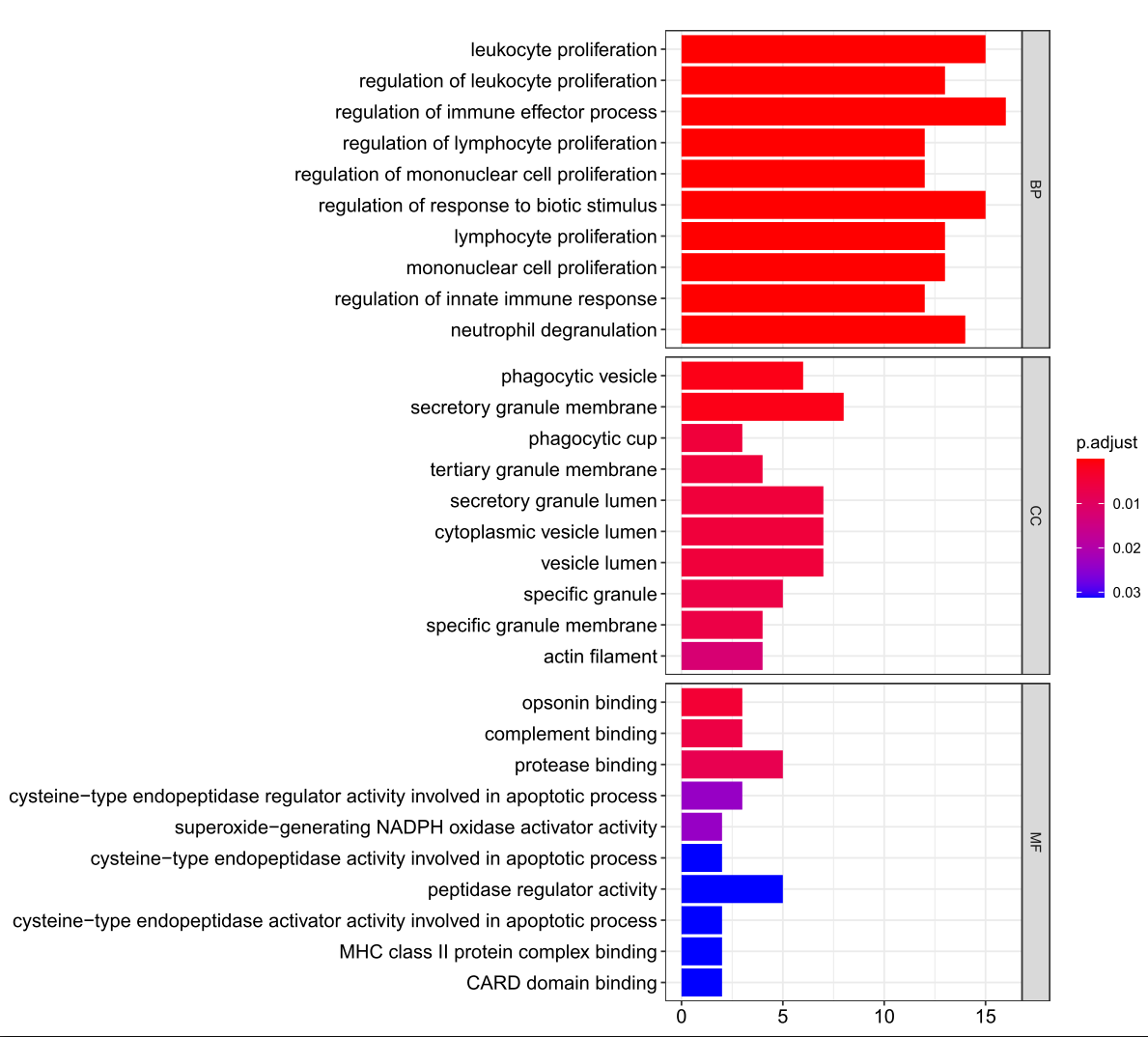
## GO 富集分析
ego <- enrichGO(
gene = gene$ENTREZID,
keyType = "ENTREZID",
OrgDb = org.Hs.eg.db,
ont = "all",
pAdjustMethod = "BH",
pvalueCutoff = 0.05,
readable = TRUE)
pdf(file = "GO.pdf", width = 10, height = 9)
barplot(ego, drop = TRUE, showCategory = 10,split="ONTOLOGY") +
scale_y_discrete(labels=function(x) str_wrap(x, width=80))+
facet_grid(ONTOLOGY~., scale='free')
dev.off()
#
dotplot(ego, showCategory = 10)
# 导出GO数据
write.csv(ego, "05.GO.result.csv")
## KEGG 富集分析
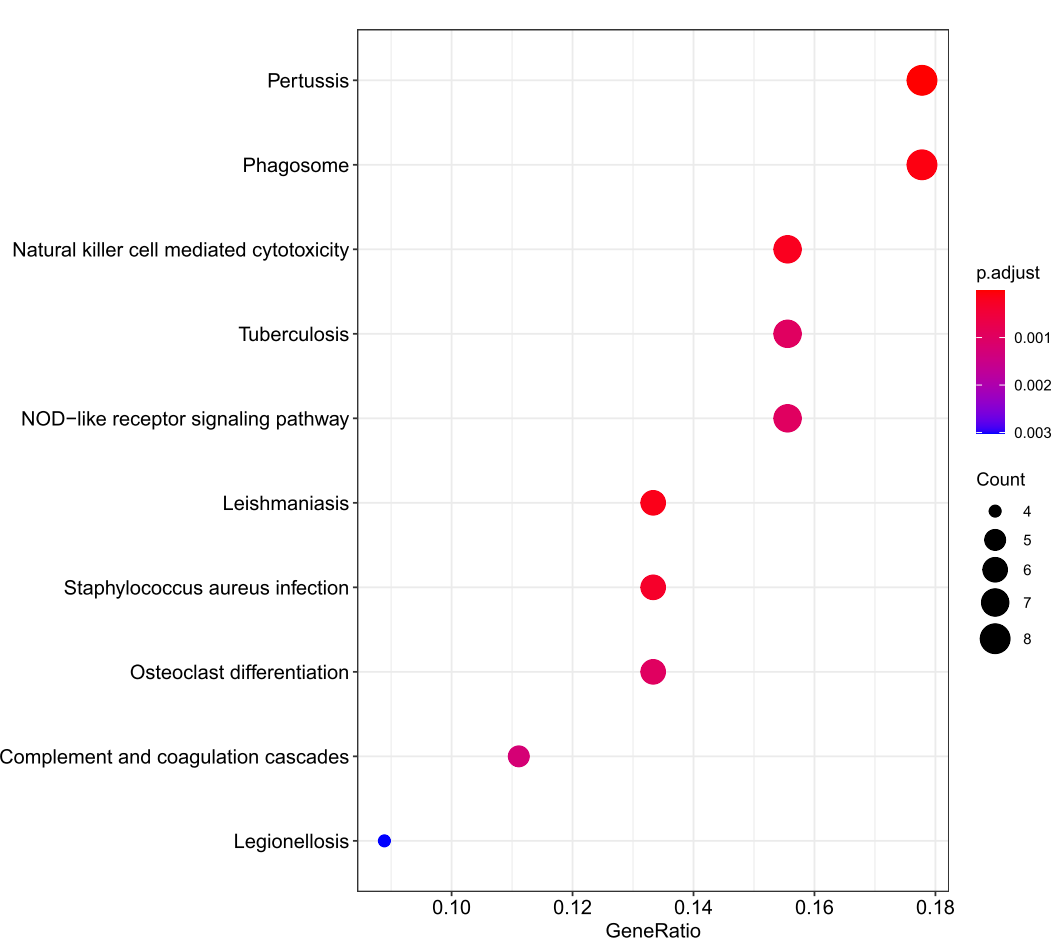
ekegg <- enrichKEGG(
gene = gene$ENTREZID,
keyType = "kegg",
organism = 'hsa',
pvalueCutoff = 0.05,
pAdjustMethod = "BH")
pdf(file = "KEGG_bar.pdf", width = 9, height = 8)
barplot(ekegg, showCategory = 10)
dev.off()
pdf(file = "KEGG_dotplot.pdf", width = 9, height = 8)
dotplot(ekegg, showCategory = 10)
dev.off()
## 导出KEGG富集分析数据
write.csv(ekegg, "06.KEGG.result.csv")

往期文章:
1. 复现SCI文章系列专栏
2. 《生信知识库订阅须知》,同步更新,易于搜索与管理。
3. 最全WGCNA教程(替换数据即可出全部结果与图形)
4. 精美图形绘制教程
5. 转录组分析教程
一个转录组上游分析流程 | Hisat2-Stringtie
小杜的生信筆記,主要发表或收录生物信息学的教程,以及基于R的分析和可视化(包括数据分析,图形绘制等);分享感兴趣的文献和学习资料!!
















 2534
2534











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











