







RNA Velocity 分析流程笔记
🔹 什么是 RNA Velocity?
RNA velocity 是一种通过单细胞 RNA 测序(scRNA-seq)数据推测细胞未来状态的分析方法。它基于细胞中spliced(剪切后)和 unspliced(未剪切)mRNA的比例,来计算基因表达变化的速率,并预测细胞的发育轨迹。
这一方法最早由La Manno et al., 2018在 Nature 发表:
**La Manno, G., Soldatov, R., Zeisel, A. et al. RNA velocity of single cells.**Nature560, 494–498 (2018).DOI: 10.1038/s41586-018-0414-6
🔹 RNA Velocity 解决的问题
- 预测细胞命运:基于 mRNA 的 splicing 动态,推测细胞在未来的转录状态。
- 揭示发育轨迹:用于分析干细胞、分化细胞如何随时间变化。
- 探索细胞状态转换:识别具有不同分化潜力的细胞群。
- 优化细胞聚类分析:结合传统 UMAP/T-SNE,RNA velocity 提供更丰富的时间信息。
🔹 RNA Velocity 的应用
- 神经发育研究(La Manno et al., 2018):神经干细胞如何分化为不同亚型。
- 免疫细胞发育:揭示 T 细胞和 B 细胞在免疫应答中的动态。
- 癌症研究:研究肿瘤微环境中免疫细胞的动态变化。
- 再生医学:预测干细胞如何转变为特定组织类型。
🔹 1. 生成 .loom 文件(运行 Velocyto)
RNA velocity 依赖 Velocyto 计算 spliced/unspliced 数据,必须基于 BAM 和 GTF 文件生成 .loom 文件。
(1) 运行 Velocyto 计算 spliced/unspliced 计数
在 Cell Ranger 预处理过的单细胞数据目录(包含 filtered_feature_bc_matrix 和 possorted_genome_bam.bam)下运行:
velocyto run -b filtered_feature_bc_matrix/barcodes.tsv \
-o velocyto_output \
-m /path/to/repeat_mask.gtf \
/path/to/possorted_genome_bam.bam \
/path/to/genome_annotation.gtf
其中:
-b:指定细胞条形码文件-o:输出.loom文件的目录-m:可选的 repeat_mask.gtf 以屏蔽重复区域possorted_genome_bam.bam:Cell Ranger 生成的 BAM 文件genome_annotation.gtf:基因组 GTF 注释文件
(2) 以 Shell 脚本形式后台运行
为了避免终端关闭导致任务中断,可以将 Velocyto 命令写入 Shell 脚本,并使用 nohup 在后台运行:
**创建 Shell 脚本 **run_velocyto.sh
echo "Running Velocyto..."
velocyto run -b filtered_feature_bc_matrix/barcodes.tsv \
-o velocyto_output \
-m /path/to/repeat_mask.gtf \
/path/to/possorted_genome_bam.bam \
/path/to/genome_annotation.gtf
echo "Velocyto completed!"
后台运行 Shell 脚本
nohup bash run_velocyto.sh > velocyto_log.txt 2>&1 &
nohup:保证任务在终端关闭后仍然运行> velocyto_log.txt 2>&1:将标准输出和错误日志重定向到velocyto_log.txt&:让任务在后台运行
检查运行状态
tail -f velocyto_log.txt # 查看实时日志
(3) 确保 .loom 文件生成成功
.loom 文件将存储于 velocyto_output 目录下,例如:
ls velocyto_output/
run_sample.loom
🔹 2. 读取数据并转换格式
(1) 读取 Seurat 处理后的数据并转换为 AnnData
RNA velocity 需要基于 spliced/unspliced 数据,而 Seurat 主要处理标准化的表达矩阵。因此,我们需要将 Seurat 对象转换为 Scanpy 兼容的 AnnData 格式。
import scanpy as sc
import pandas as pd
import numpy as np
from scipy.io import mmread
# 读取计数矩阵
counts = mmread("counts.mtx").T.tocsr()
# 读取基因和细胞名称
genes = pd.read_csv("genes.csv", header=None).squeeze("columns")
cells = pd.read_csv("barcodes.csv", header=None).squeeze("columns")
# 创建 AnnData 对象
adata = sc.AnnData(X=counts)
adata.var_names = genes
adata.obs_names = cells
# 读取元数据
metadata = pd.read_csv("metadata.csv", index_col=0)
adata.obs = metadata
(2) 读取 .loom 文件以获取 spliced/unspliced 数据
RNA velocity 依赖于 velocyto 计算出的 .loom 文件,该文件包含 spliced/unspliced 层。
import scvelo as scv
# 读取 loom 文件
aadata_loom = scv.read("sample.loom")
检查细胞名称是否匹配
common_cells = list(set(adata.obs_names) & set(aadata_loom.obs_names))
adata_filtered = adata[adata.obs_names.isin(common_cells)].copy()
aadata_loom_filtered = aadata_loom[aadata_loom.obs_names.isin(common_cells)].copy()
确保基因名称也匹配
common_genes = list(set(adata_filtered.var_names) & set(aadata_loom_filtered.var_names))
adata_filtered = adata_filtered[:, common_genes].copy()
aadata_loom_filtered = aadata_loom_filtered[:, common_genes].copy()
🔹 3. 复制 spliced/unspliced 层并检查数据
adata_filtered.layers["spliced"] = aadata_loom_filtered.layers["spliced"]
adata_filtered.layers["unspliced"] = aadata_loom_filtered.layers["unspliced"]
确保 spliced/unspliced 数据为 float 类型
adata_filtered.layers["spliced"] = adata_filtered.layers["spliced"].astype(np.float32)
adata_filtered.layers["unspliced"] = adata_filtered.layers["unspliced"].astype(np.float32)
🔹 4. 计算 RNA velocity
# 1️⃣ 预处理
scv.pp.filter_and_normalize(adata_filtered, min_counts=20, n_top_genes=2000)
scv.pp.moments(adata_filtered, n_pcs=30, n_neighbors=30)
# 2️⃣ 计算 RNA velocity
scv.tl.velocity(adata_filtered, mode='stochastic')
scv.tl.velocity_graph(adata_filtered)
可视化
import matplotlib.pyplot as plt
scv.pl.velocity_embedding_stream(
adata_filtered,
basis="umap",
color="celltype",
figsize=(8, 6),
show=False # 关闭实时显示,避免重复绘制
)
# 手动保存 PDF
plt.savefig("../output_data/velocity_embedding_celltype.pdf",
dpi=300, bbox_inches="tight", format="pdf")
plt.show()
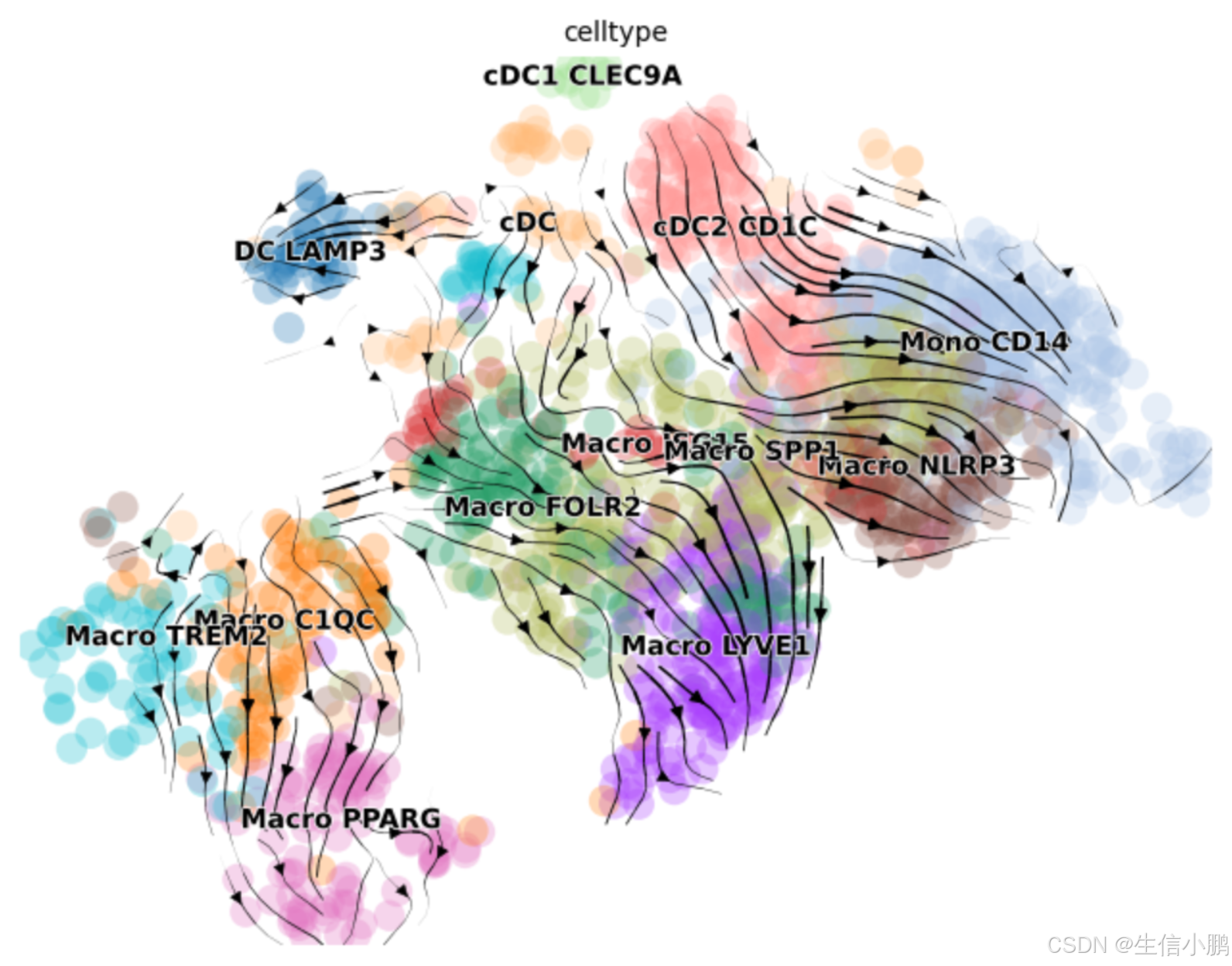
scv.pl.scatter(adata_filtered, basis="umap", color=["ITGB1", "SOX2", "TP53"])
🔹 5. 计算 latent time
scv.tl.recover_dynamics(adata_filtered)
scv.tl.velocity(adata_filtered, mode="dynamical")
scv.tl.velocity_graph(adata_filtered)
scv.tl.latent_time(adata_filtered)
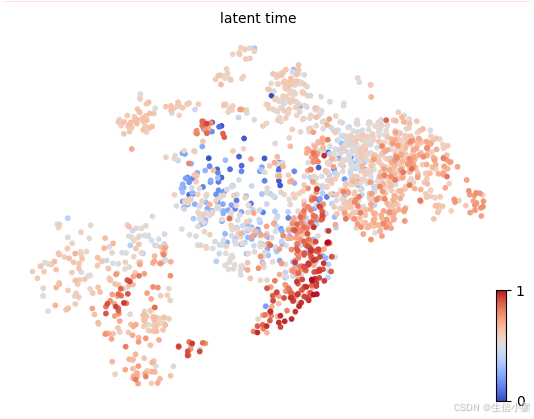
可视化 latent time:
scv.pl.scatter(adata_filtered, color="latent_time", cmap="coolwarm")
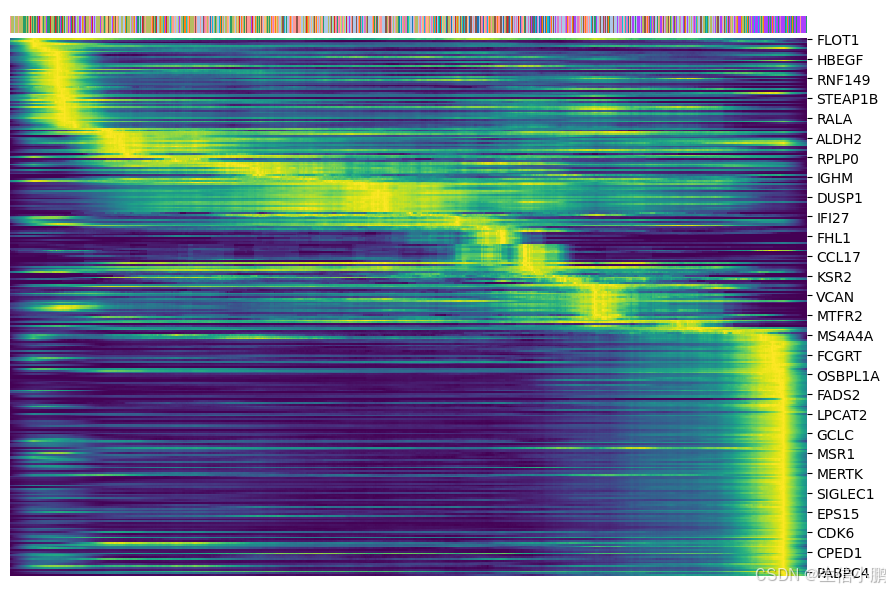
基因筛选并绘制热图
top_genes = adata_filtered.var['fit_likelihood'].sort_values(ascending=False).index[:300]
scv.pl.heatmap(adata_filtered, var_names=top_genes, sortby='latent_time', col_color='cell_type', n_convolve=100)
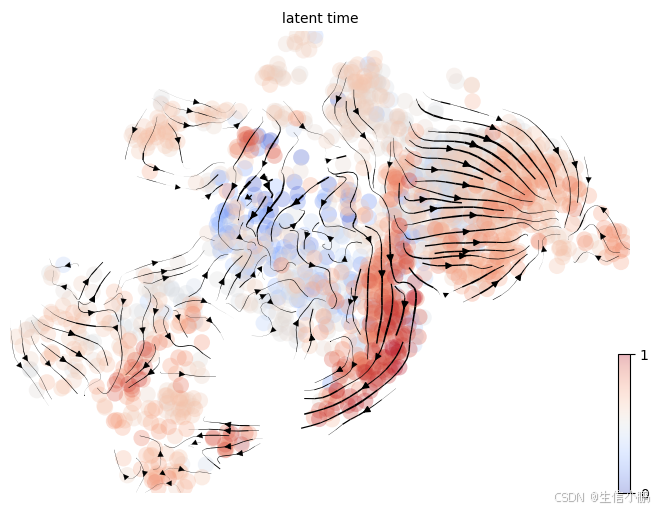
🔹 6. RNA velocity 结果可视化
流场图(velocity embedding stream)
scv.pl.velocity_embedding_stream(adata_filtered, basis="umap", color="latent_time", cmap="coolwarm", figsize=(8, 6))
矢量图(velocity embedding)
scv.pl.velocity_embedding(adata_filtered, basis="umap", arrow_size=1.5, arrow_length=3)

单基因 velocity
scv.pl.velocity(adata_filtered, ['MYC', 'SOX2', 'TP53'])
🔹 7. 结果保存
保存高分辨率图片
import matplotlib.pyplot as plt
scv.pl.velocity_embedding_stream(adata_filtered, basis="umap", color="latent_time", show=False)
plt.savefig("../output/latent_time_velocity.pdf", dpi=300, bbox_inches="tight", format="pdf")
plt.show()
保存 AnnData 结果
adata_filtered.write("../output/adata_velocity.h5ad")
🔹 结论总结
RNA velocity 是一种强大的单细胞转录组分析工具,它可以预测细胞的未来状态,揭示发育轨迹,并用于探索细胞状态转换。通过Velocyto 计算 spliced/unspliced 数据,并结合Scanpy 和 scVelo 进行分析,我们能够获得细胞动态变化的丰富信息。
✅ 关键步骤回顾
- 数据预处理:从 Seurat 转换数据,并匹配
.loom文件。 - RNA velocity 计算:利用
scVelo计算stochastic或dynamicalvelocity。 - Latent time 分析:预测细胞发育时间,并寻找关键基因。
- 可视化与解释:使用 UMAP、stream plots 和 heatmaps 展示 velocity 轨迹。
- 结果应用:结合生物学背景,探索免疫细胞、癌症研究等领域中的动态变化。
🚀**RNA velocity 提供了一个全新的角度来理解细胞的动态变化,为单细胞转录组分析带来了更深入的时空信息!**🎉


















 3246
3246

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











