







如何获取 genome_annotation.gtf 和 repeat_mask.gtf 及确定 BAM 文件匹配的基因组版本
在单细胞分析(16)—— RNA Velocity 分析流程中提及了运行RNA Velocity的两个关键文件,genome_annotation.gtf和repeat_mask.gtf,
velocyto run -b filtered_feature_bc_matrix/barcodes.tsv \
-o velocyto_output \
-m /path/to/repeat_mask.gtf \
/path/to/possorted_genome_bam.bam \
/path/to/genome_annotation.gtf
其中:
-b:指定细胞条形码文件-o:输出.loom文件的目录-m:可选的 repeat_mask.gtf 以屏蔽重复区域possorted_genome_bam.bam:Cell Ranger 生成的 BAM 文件genome_annotation.gtf:基因组 GTF 注释文件
关于genome_annotation.gtf和repeat_mask.gtf这两个文件的有关内容和下载如下。
🔹 GTF 文件的内容和用途
GTF(Gene Transfer Format)文件是用于基因组注释的标准格式,主要包含基因、转录本、外显子和其他功能元件的信息。它们用于单细胞 RNA 测序(scRNA-seq)分析,如RNA velocity、基因表达定量和功能注释。
**genome_annotation.gtf**(基因组注释 GTF 文件)- 存储内容:基因坐标、转录本结构、外显子和内含子信息。
- 用途:
- 确定 RNA 片段的基因来源。
- 计算spliced/unsplicedmRNA 比例。
- 用于RNA velocity和转录本组装。
**repeat_mask.gtf**(重复序列屏蔽 GTF 文件)- 存储内容:基因组中的重复序列信息,如转座子和简单重复序列。
- 用途:
- 过滤掉低质量或误匹配的 reads。
- 避免重复序列影响 RNA velocity 计算。
- 确保数据分析的准确性,减少假阳性信号。
🔹 获取 genome_annotation.gtf(基因组注释 GTF 文件)
genome_annotation.gtf 是标准的基因组 GTF 文件,包含所有已知基因的 外显子(exon)信息,用于 Velocyto 计算 spliced 片段。
✅ 方法 1:从 GENCODE/Ensembl 下载
你可以从 GENCODE 或 Ensembl 下载 符合你物种的基因组 GTF 文件。
1️⃣ GENCODE 下载(推荐)

- 人类 (GRCh38):
wget ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_43/gencode.v43.annotation.gtf.gz
gunzip gencode.v43.annotation.gtf.gz
- 小鼠 (GRCm39):
wget ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_mouse/release_M32/gencode.vM32.annotation.gtf.gz
gunzip gencode.vM32.annotation.gtf.gz
2️⃣ Ensembl 下载
wget ftp://ftp.ensembl.org/pub/release-110/gtf/homo_sapiens/Homo_sapiens.GRCh38.110.gtf.gz
gunzip Homo_sapiens.GRCh38.110.gtf.gz
💡 注意:
- Ensembl 的 GTF 文件格式与 GENCODE 类似,适用于大多数物种。
- 确保下载 与 BAM 文件匹配的参考基因组版本(如
GRCh38或GRCm39)。
🔹 获取 repeat_mask.gtf(重复序列屏蔽 GTF 文件)
repeat_mask.gtf 主要用于过滤 重复序列(repeat elements),避免错误地归类 RNA velocity 数据。通常由 RepeatMasker 生成。
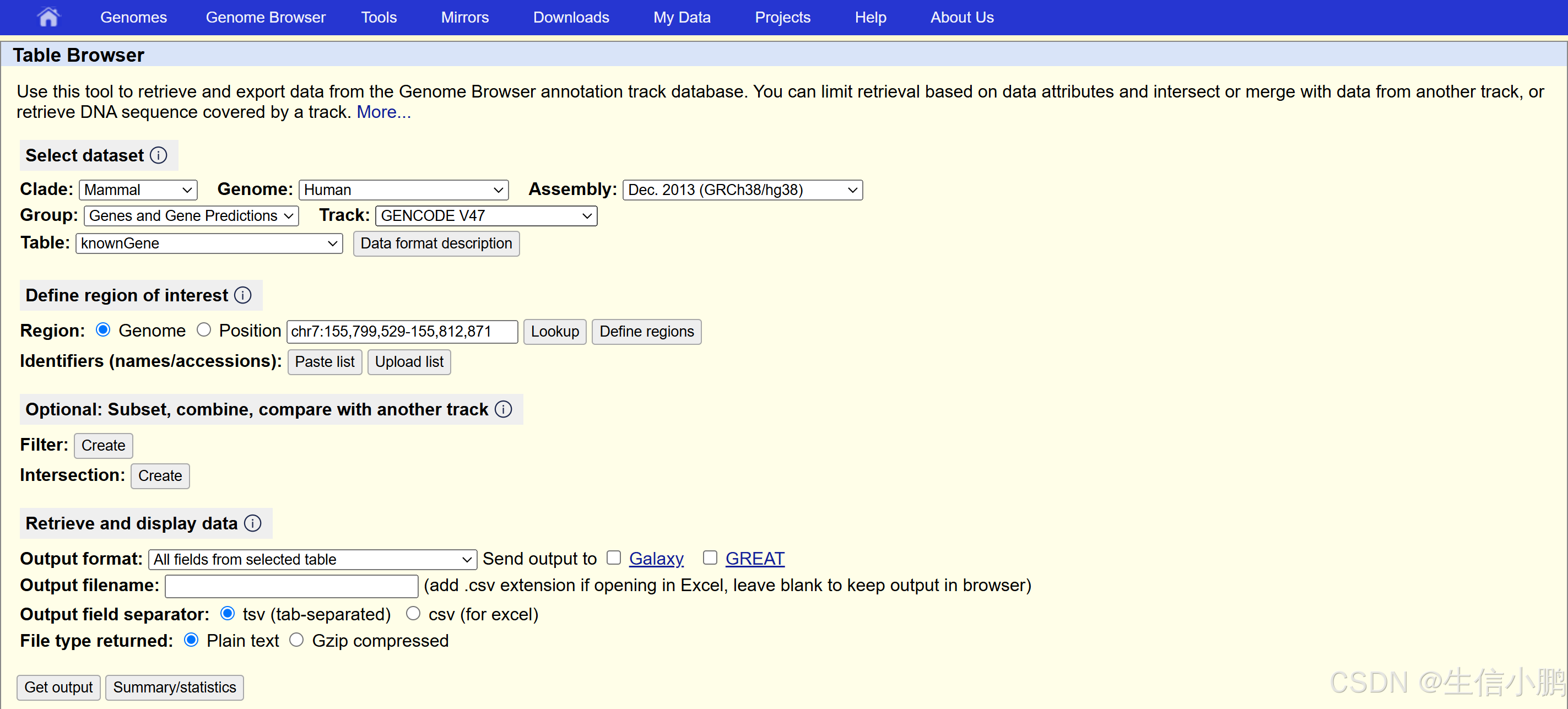
✅ 方法 1:从 UCSC Table Browser 下载(推荐)
- 进入 UCSC Table Browser
- 选择你的基因组版本(如 GRCh38/hg38)
- 选择 Track:
RepeatMasker - 选择 Output Format:
GTF - **点击 “Get Output” 下载 **
repeat_mask.gtf
✅ 方法 2:从 UCSC FTP 服务器下载
wget http://hgdownload.soe.ucsc.edu/goldenPath/hg38/database/rmsk.txt.gz
gunzip rmsk.txt.gz
然后转换为 GTF 格式:
cut -f 6-9,12,14,15,16,11 rmsk.txt | awk 'BEGIN{OFS="\t"} {print $1,"RepeatMasker","exon",$2,$3,".",$4,".","gene_id \""$5"\"; transcript_id \""$5"\";"}' > repeat_mask.gtf
✅ 方法 3:使用 RepeatMasker 生成
RepeatMasker -species human -gff genome.fa
然后提取 repeat_mask.gtf:
grep -v "#" genome.fa.out | awk '{print $5, "RepeatMasker", "exon", $6, $7, ".", $9, ".", "gene_id \"repeat_"$10"\"; transcript_id \"repeat_"$10"\";"}' > repeat_mask.gtf
🔹 如何确定 BAM 文件的匹配基因组版本?
✅ 方法 1:使用 samtools 查看 BAM 头信息
samtools view -H your_file.bam | grep "@SQ"
示例输出:
@SQ SN:chr1 LN:248956422
@SQ SN:chr2 LN:242193529
...
@HD VN:1.6 SO:coordinate
@PG ID:STAR PN:STAR VN:2.7.10a
@CO Genome Reference: GRCh38.primary_assembly.genome.fa
关键点:
@SQ:染色体信息,SN:chr1等表示染色体编号。@CO Genome Reference(如果存在):直接告诉你参考基因组名称,如GRCh38.primary_assembly.genome.fa。
✅ 方法 2:使用 samtools idxstats 确认染色体名称
samtools idxstats your_file.bam
chr1, chr2, chrX→ UCSC 参考基因组(hg19/hg38)。1, 2, X→ Ensembl 参考基因组(GRCh37/GRCh38)。
✅ 方法 3:使用 bamtools 检查 BAM 头信息
bamtools header -in your_file.bam
如果 BAM 头中包含:
@CO Genome Reference: Homo_sapiens.GRCh38.dna.primary_assembly.fa
则说明该 BAM 文件使用的是 GRCh38 版本。
✅ 方法 4:检查 BAM 生成工具
samtools view -H your_file.bam | grep "@PG"
示例输出:
@PG ID:STAR PN:STAR VN:2.7.3a CL:STAR --genomeDir /path/to/genome_GRCh38
--genomeDir指向GRCh38,则 BAM 参考基因组是 GRCh38。--genomeDir指向mm10,则 BAM 参考基因组是 小鼠 GRCm38。
🔹 结论总结
| 文件 | 作用 | 推荐下载 | 备用方案 |
|---|---|---|---|
| genome_annotation.gtf | 提供基因注释(外显子信息) | GENCODE / Ensembl | 手动提取 GTF |
| repeat_mask.gtf | 屏蔽重复序列,避免错误归类 | UCSC Table Browser | UCSC FTP / RepeatMasker |
🚀 使用这些方法,就能正确获取 genome_annotation.gtf 和 repeat_mask.gtf,并确认 BAM 文件的基因组版本! 🎉


















 416
416

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











