






1. macs3识别
macs3 callpeak -c SRR15567099.bam -t SRR13755437.bam -p 1e-10 -f BAM -g hs -n HMEC_rep1 --call-summits --outdir ./output
macs3 callpeak -c SRR15567100.bam -t SRR13755438.bam -p 1e-10 -f BAM -g hs -n HMEC_rep2 --call-summits --outdir ./output
识别得到的结果:

查看
wc -l HMEC_rep1_peaks.narrowPeak
wc -l HMEC_rep2_peaks.narrowPeak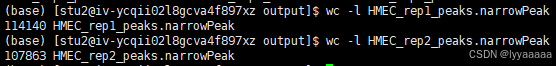
2. bedtools合并生物重复
bedtools intersect -a HMEC_rep1_peaks.narrowPeak -b HMEC_rep2_peaks.narrowPeak -f 0.50 > hmec_both_peaks.bed
sort -k1,1 -k2,2n -k7,7nr HMEC_both_peaks.bed | awk '!seen[$1$2$3]++' > filtered_peaks.bed
bedtools intersect -a filtered_peaks.bed -b /student2/graduate/result/all_promoter.bed -v > peaks_without_promoters.bed
awk '{print $1"\t"$2"\t"$3"\t"$7}' peaks_without_promoters.bed > peaks_without_promoters.bedGraph-f 0.75:设置重叠至少达到75%


















 900
900

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









