






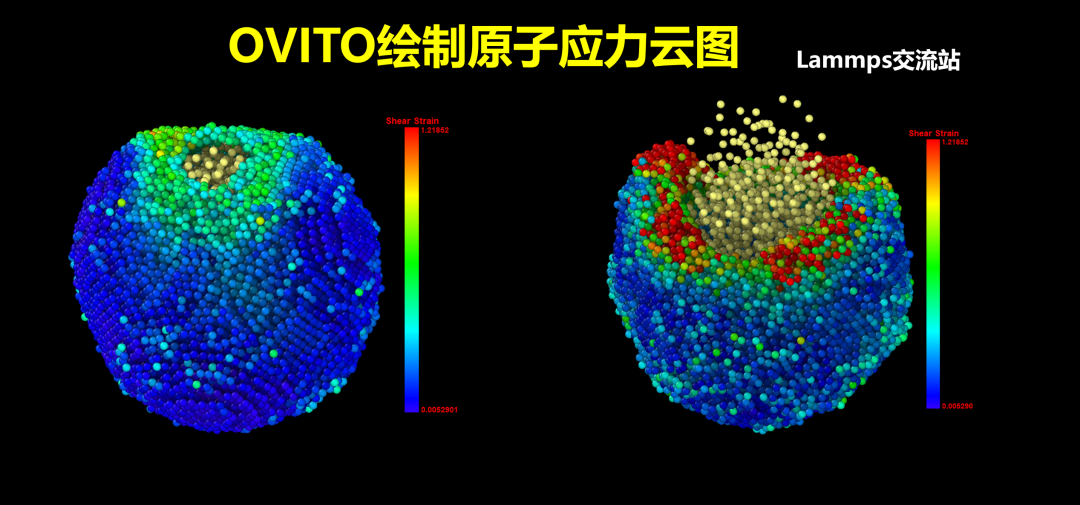
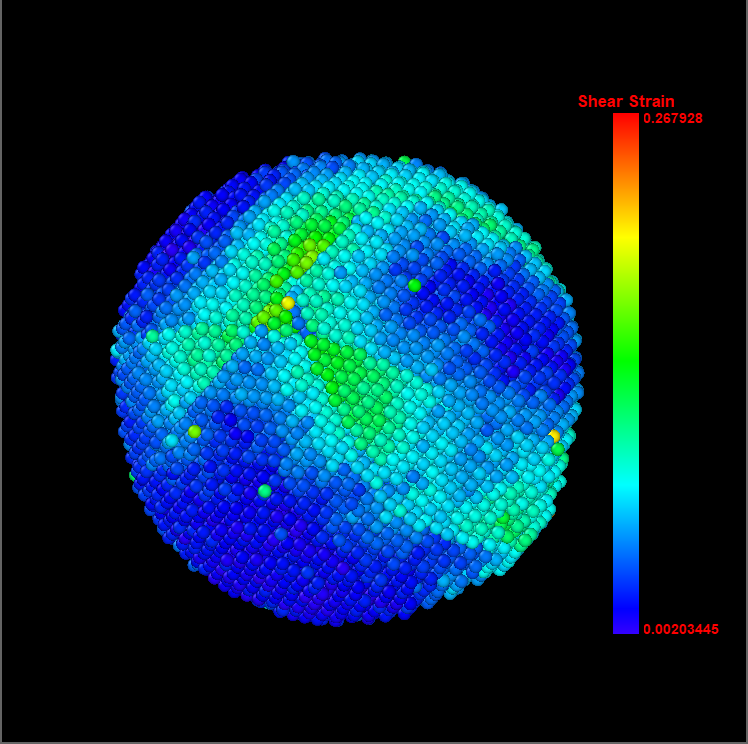
在我们用分子动力学模拟力学性能时,应力应变云图是我们模拟结果中常常出现的,如下图为分子动力学的铜铅合金拉伸变形特性的研究一文中铜铅合金剪应力云图,本文介绍如何利用OVITO软件绘制原子应力云图。
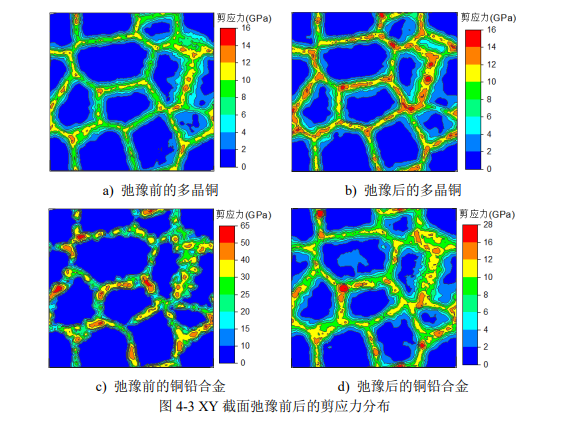
图参考自:韩浏淼. 基于分子动力学的铜铅合金拉伸变形特性的研究[D].哈尔滨工业大学,2018.

具体步骤
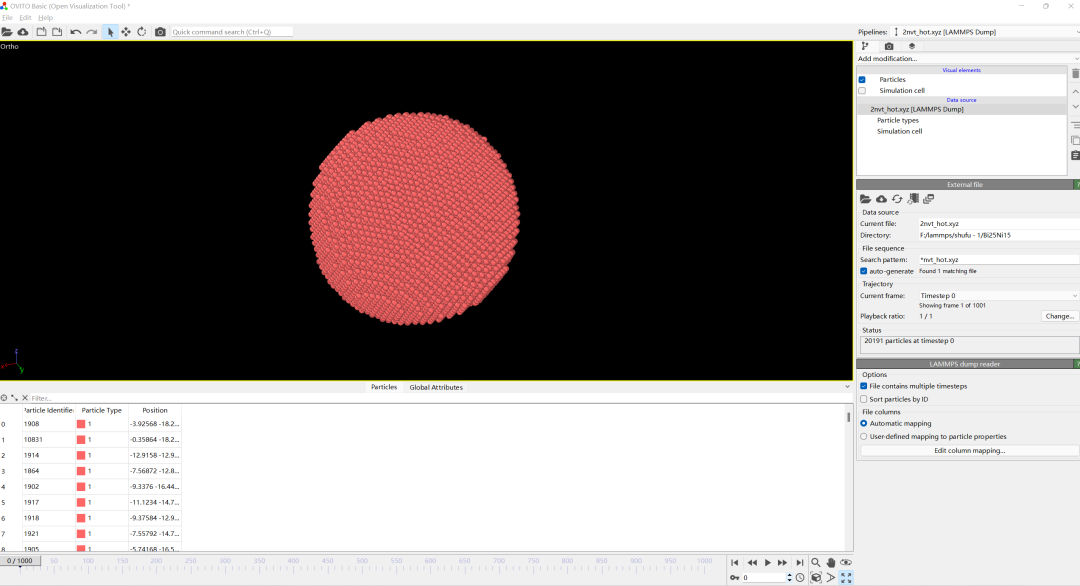
1.导入dump文件
在OVITO软件中导入我们的结果dump文件。
2.Add modification--Atomic strain
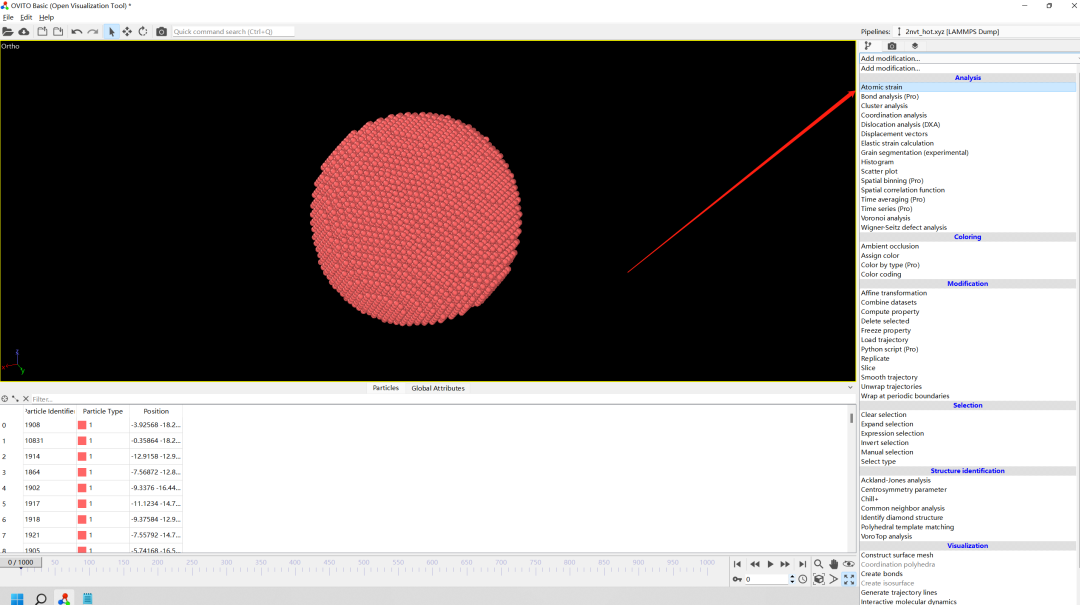
3.Add modification--Atomic strain
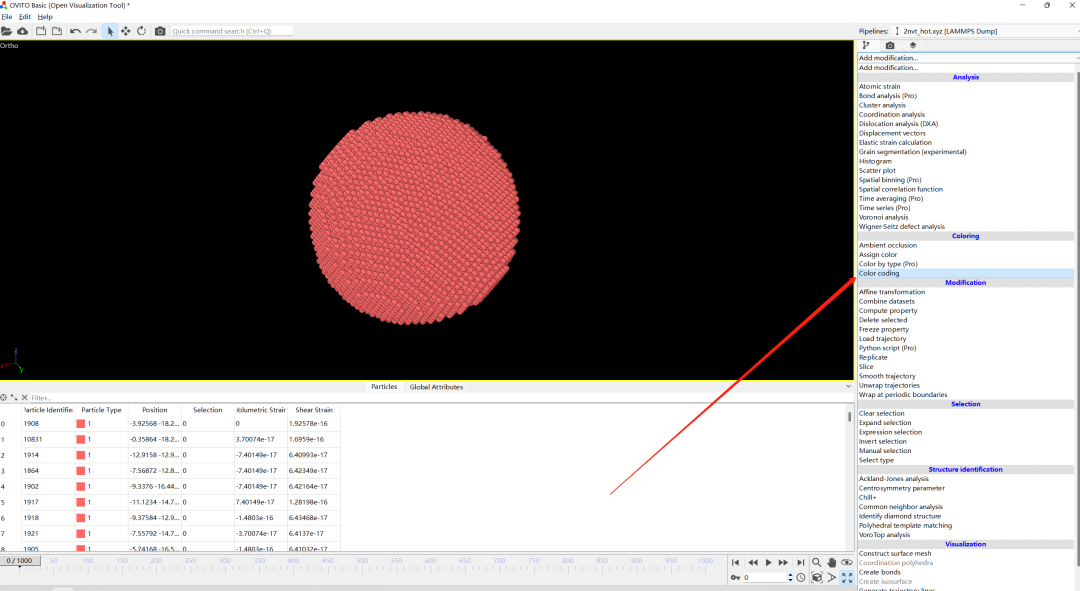
4.选择我们需要绘制的云图类型
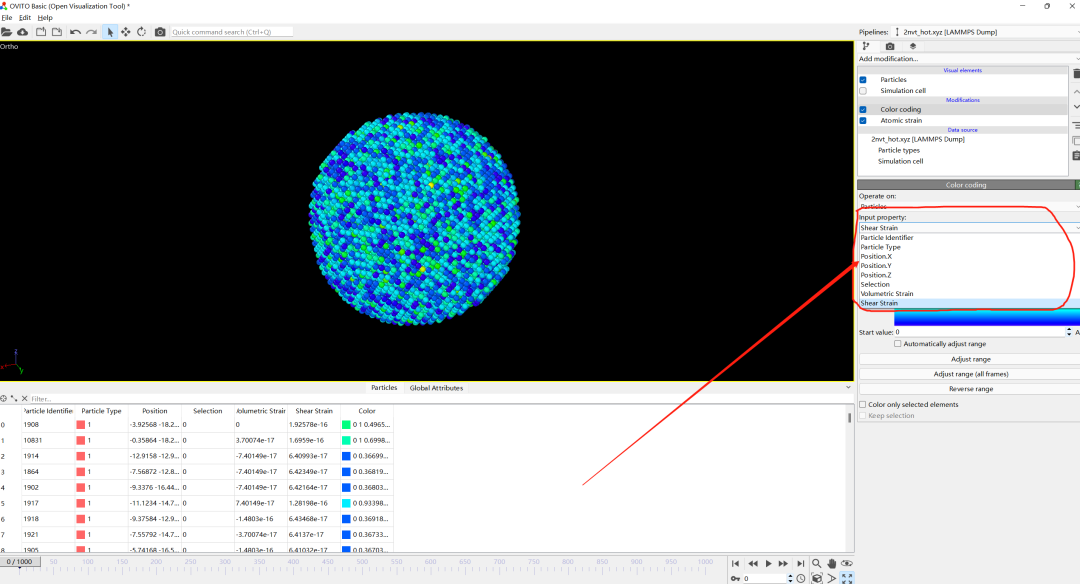
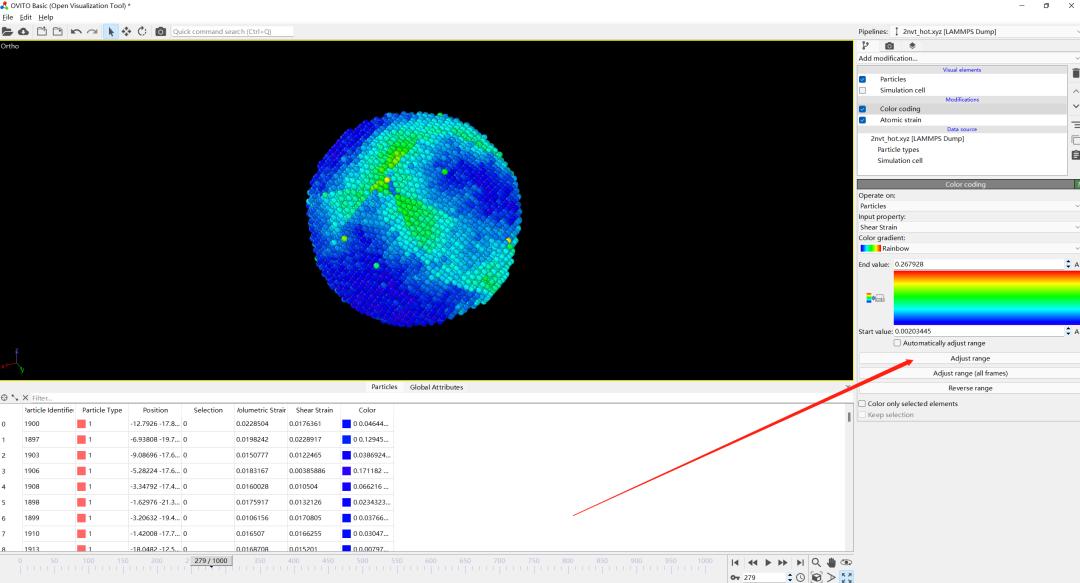
5.Adjust range
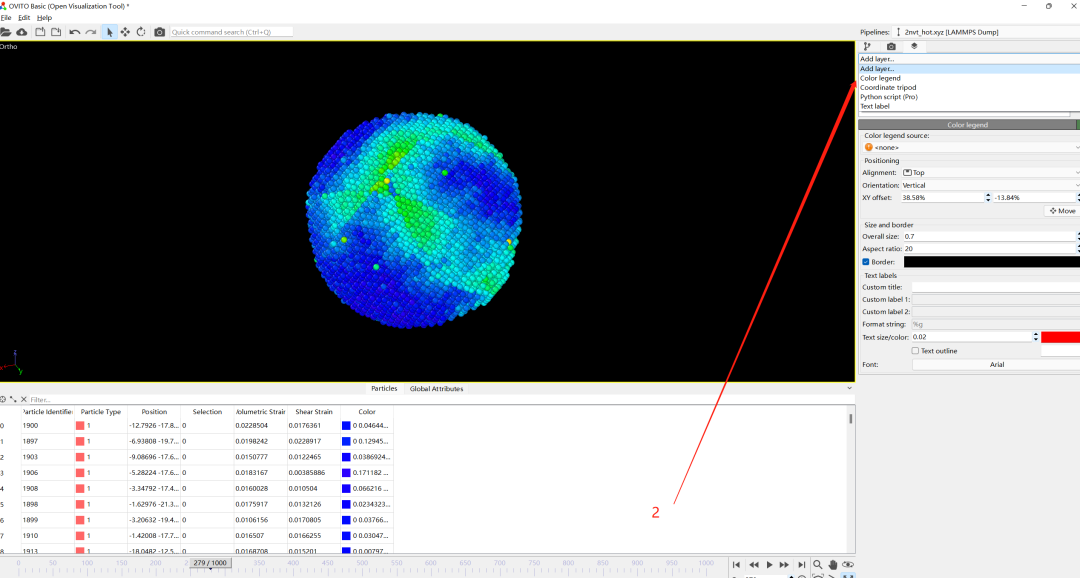
6.添加标尺
1)add layer
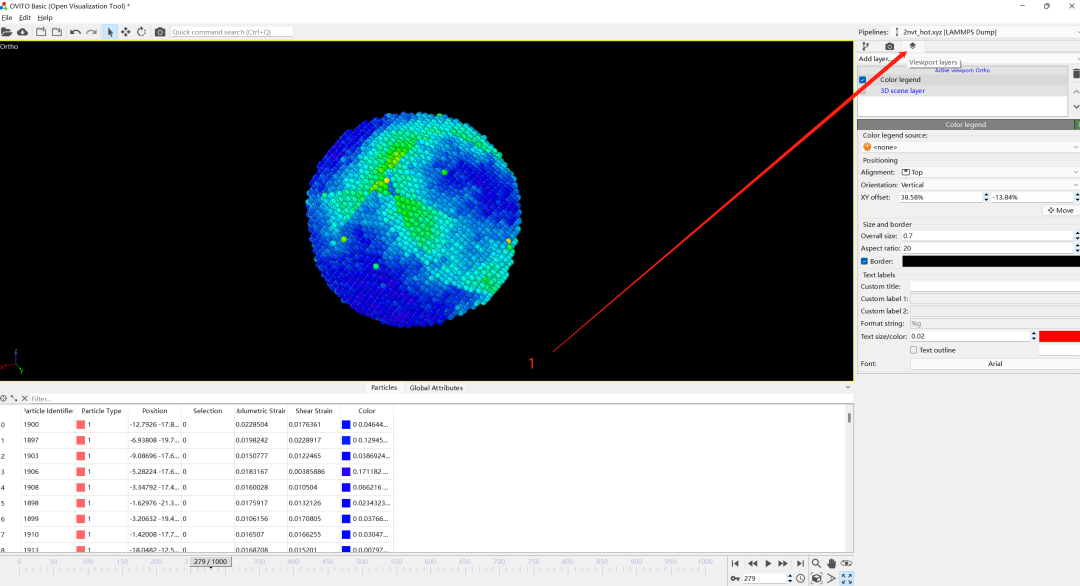
2) Color legend


更多案例请关注 公众号:lammps交流站

















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









