







先自我介绍一下,小编浙江大学毕业,去过华为、字节跳动等大厂,目前阿里P7
深知大多数程序员,想要提升技能,往往是自己摸索成长,但自己不成体系的自学效果低效又漫长,而且极易碰到天花板技术停滞不前!
因此收集整理了一份《2024年最新Linux运维全套学习资料》,初衷也很简单,就是希望能够帮助到想自学提升又不知道该从何学起的朋友。

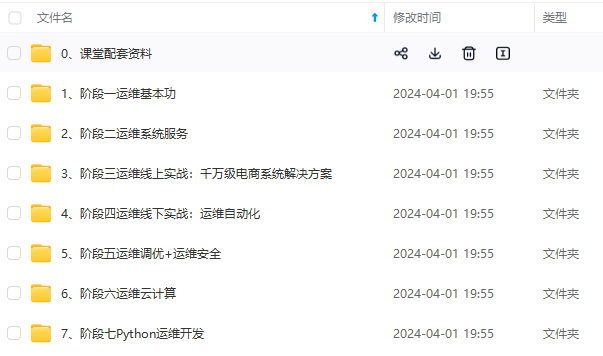
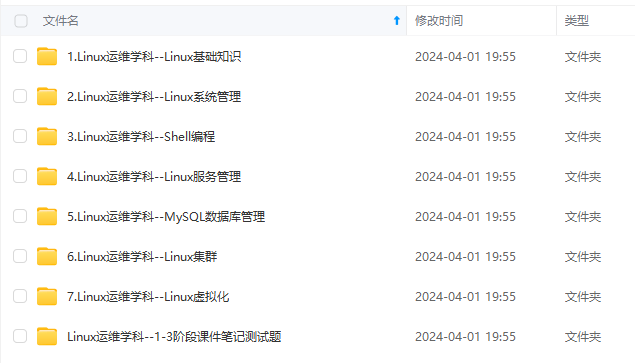
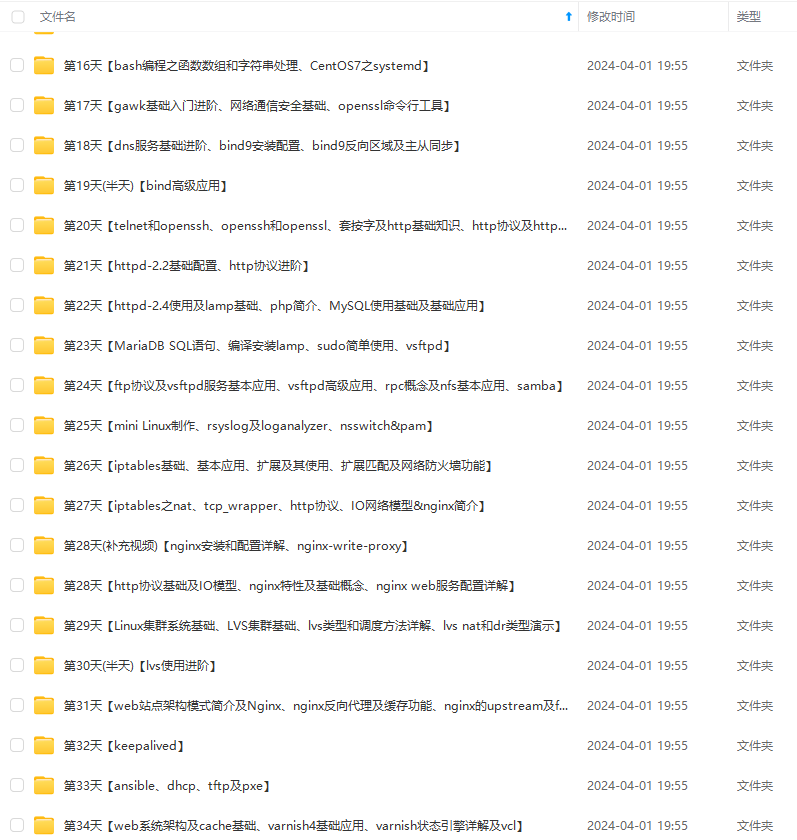
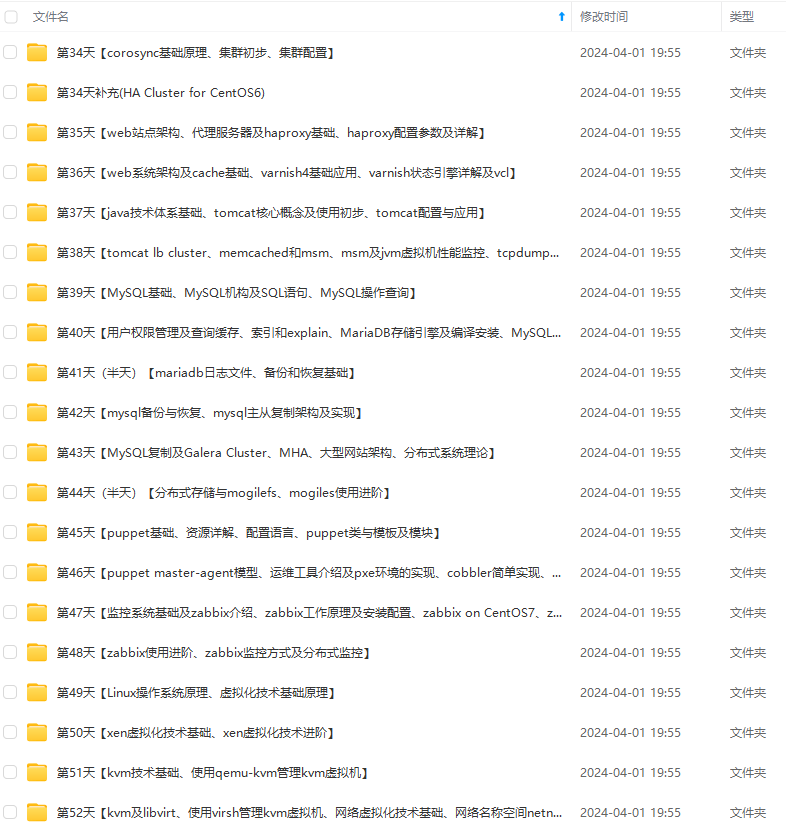
既有适合小白学习的零基础资料,也有适合3年以上经验的小伙伴深入学习提升的进阶课程,涵盖了95%以上运维知识点,真正体系化!
由于文件比较多,这里只是将部分目录截图出来,全套包含大厂面经、学习笔记、源码讲义、实战项目、大纲路线、讲解视频,并且后续会持续更新
如果你需要这些资料,可以添加V获取:vip1024b (备注运维)

正文
###与checkV和DRAMv一起安装,整合基因注释,单独使用virsorter的就可以直接略过。
install VirSorter2, checkV and DRAMv
conda create -n viral-id-sop virsorter=2 checkv dram
activate env
conda activate viral-id-sop
vs2 db: db-vs2
virsorter setup -d db-vs2 -j 4
checkv db: checkv-db-v1.0
checkv download_database .
DRAMv: db-dramv
DRAM-setup.py prepare_databases --skip_uniref --output_dir db-dramv
########################################################33333
##以下是官方建议的SOP分析流程
#Run VirSorter2
#First, run VirSorter2 with a loose cutoff of 0.5 for maximal sensitivity. We are only interested in phages (dsDNA and ssDNA phage). A minimal length 5000 bp is chosen since it is the minimum required by downstream viral
classification. You can adjust the “-j” option based on the availability of CPU cores. Note that the “–keep-original-seq” option preserves the original sequence of circular and (near) fully viral contigs (score >0.8 as a whole sequence) and we are passing them to checkV to trim possible host genes left at ends and handle duplicate segments of circular contigs.
virsorter run --keep-original-seq -i 5seq.fa -w vs2-pass1 --include-groups dsDNAphage,ssDNA --min-length 5000 --min-score 0.5 -j 28 all
#Run checkV
#There could be some non-viral sequences or regions in the VirSorter2 results with a minimal score cutoff of 0.5. Here we use CheckV to quality control the VirSorter2 results and also to trim potential host regions left at the ends of proviruses. You can adjust the “-t” option based on the availability of CPU cores.
checkv end_to_end vs2-pass1/final-viral-combined.fa checkv -t 28 -d /fs/project/PAS1117/jiarong/db/checkv-db-v1.0
#Run VirSorter2 again
#Then we run the checkV-trimmed sequences through VirSorter2 again to generate “affi-contigs.tab” files needed by DRAMv to identify AMGs. You can adjust the “-j” option based on the availability of CPU cores. Note the “–seqname-suffix-off” option preserves the original input sequence name since we are sure there is no chance of getting >1 proviruses from the same contig in this second pass, and the “–viral-gene-enrich-off” option turns off the requirement of having more viral genes than host genes to make sure that VirSorter2 is not doing any screening at this step. The above two options require VirSorter2 version >=2.2.1.
cat checkv/proviruses.fna checkv/viruses.fna > checkv/combined.fna
virsorter run --seqname-suffix-off --viral-gene-enrich-off --prep-for-dramv -i checkv/combined.fna -w vs2-pass2 --include-groups dsDNAphage,ssDNA --min-length 5000 --min-score 0.5 -j 28 all
#Run DRAMv
#Then run DRAMv to annotate the identified sequences, which can be used for manual curation. You can adjust the “–threads” option based on availability of CPU cores.
step 1 annotate
DRAM-v.py annotate -i vs2-pass2/for-dramv/final-viral-combined-for-dramv.fa -v vs2-pass2/for-dramv/viral-affi-contigs-for-dramv.tab -o dramv-annotate --skip_trnascan --threads 28 --min_contig_size 1000
#step 2 summarize anntotations
DRAM-v.py distill -i dramv-annotate/annotations.tsv -o dramv-distill
### 手动安装开发版,就是最新功能版
mamba create -n vs2 -c conda-forge -c bioconda “python>=3.6,<=3.10” scikit-learn=0.22.1 imbalanced-learn pandas seaborn hmmer==3.3 prodigal screed ruamel.yaml “snakemake>=5.18,<=5.26” click “conda-package-handling<=1.9”
mamba activate vs2
git clone https://github.com/jiarong/VirSorter2.git
cd VirSorter2
pip install -e .
github访问不顺畅的可以直接在这里下载Virsorter2的最新版,[https://download.csdn.net/download/zrc\_xiaoguo/88571289?spm=1001.2014.3001.5501]( )
## 2、配置数据库
### 自动配置
###新安装的情况下直接使用命令会自动下载和配置数据库到db目录下,db路径建议使用绝对路径,-j是线程数,自己指定
virsorter setup -d db -j 4
###如果不是第一次,或是之前失败了就得先删除然后重新下载和配置
rm -rf db
virsorter setup -d db -j 4
### 手动配置
###先下载对应数据库文件,这个直接浏览器回车就会下载文件出现db.tgz:
https://osf.io/v46sc/download
tar -xzf db.tgz
virsorter config --init-source --db-dir=./db
## 3、工具使用
注意事项,这个要说一下,以下是官方的说明,也就是最好使用CheckV处理序列文件去除被预测病毒区可能潜在的宿主基因
VirSorter2 tends to sometimes overestimate the size of viral sequence during provirus extraction procedure in order to achieve better sensitity. We recommend cleaning these provirus predictions to remove potential host genes on the edge of the predicted viral region, e.g. using a tool like CheckV ([Bitbucket]( )).
### 快速命令
#获取快速测试文件,当然大家可以用自己的序列文件测试
wget -O test.fa https://raw.githubusercontent.com/jiarong/VirSorter2/master/test/8seq.fa
#激活vs2环境后使用-j 4个线程运行,输入all 所有结果,-w指定输入结果的文件夹
virsorter run -w test.out -i test.fa --min-length 1500 -j 4 all
#查看结果文件夹
ls test.out
###结果文件夹中的文件主要是final-viral-combined.fa,final-viral-score.tsv,final-viral-boundary.tsv这三个文件,以下是官方介绍
Due to the large HMM database that VirSorter2 uses, this small dataset takes a few mins to finish. In the output directory (test.out), three files are useful:
final-viral-combined.fa: identified viral sequences
final-viral-score.tsv: table with score of each viral sequences across groups and a few more key features, which can be used for further filtering
final-viral-boundary.tsv: table with boundary information; This is a intermediate file that 1) might have extra records compared to other two files and should be ignored; 2) do not include the viral sequences w/ < 2 gene but have >= 1 hallmark gene; 3) the group and trim_pr are intermediate results and might not match the max_group and max_score respectively in final-viral-score.tsv
## 质量控制:
The default score cutoff (0.5) works well known viruses (RefSeq). For the real environmental data, we can expect to get false positives (non-viral) with the default cutoff. Generally, samples with more host (e.g. bulk metaG) and unknown sequences (e.g. soil) tends to have more false positives. We find a score cutoff of 0.9 work well as a cutoff for high confidence hits, but there are also many viral hits with score <0.9. It's difficult to separate the viral and non-viral hits by score alone. So we recommend using the default score cutoff (0.5) for maximal sensitivity and then applying a quality checking step using checkV. Here is a tutorial of [viral identification SOP]( ) used in Sullivan Lab.
## 更多参数设置说明:
### choosing viral groups (`--include-groups`)
The available options are dsDNAphage, NCLDV, RNA, ssDNA virus, and *lavidaviridae*. The default is dsDNAphage and ssDNA (changed from all groups since version 2.2), suitable for those only interested in phage. If you are only interested in RNA virus, you can run:
rm -rf test.out
virsorter run -w test.out -i test.fa --include-groups RNA -j 4 all
### re-run with different score cutoff (`--min-score` and `--classify`)
VirSorter2 takes one positional argument, `all` or `classify`. The default is `all`, which means running the whole pipeline, including 1) preprocessing, 2) annotation (feature extraction), and 3) classification. The main computational bottleneck is the annotation step, taking about 95% of CPU time. In case you just want to re-run with different score cutoff (--min-score), `classify` argument can skip the annotation steps, and only re-run only the classify step.
virsorter run -w test.out -i test.fa --include-groups “dsDNAphage,ssDNA” -j 4 --min-score 0.8 classify
The above overwrites the previous final output files. If you want to keep previous results, you can use `--label` to add a prefix to the new final output files.
virsorter run -w test.out -i test.fa --include-groups “dsDNAphage,ssDNA” -j 4 --min-score 0.9 --label rerun classify
### speed up a run (`--provirus-off`)
In case you need to have some results quickly, there are two options: 1) turn off provirus step with `--provirus-off`; this reduces sensitivity on sequences that are only partially viral; 2) subsample ORFs from each sequence with `--max-orf-per-seq`; This option subsamples ORFs if a sequence has more ORFs than the number provided. Note that this option is only availale when `--provirus-off` is used.
rm -rf test.out
virsorter run -w test.out -i test.fa --provirus-off --max-orf-per-seq 20 all
### 其他选项
You can run `virsorter run -h` to see all options. VirSorter2 is a wrapper around [snakemake]( ), a great pipeline management tool designed for reproducibility, and running on computer clusters. All snakemake options still work with VirSorter2, and users can simply append those snakemake option to virsorter options (after `all` or `classify`). For example, the `--forceall` snakemake option can be used to re-run the pipeline.
virsorter run -w test.out -i test.fa --provirus-off --max-orf-per-seq 20 all --forceall
When you re-run any VirSorter2 command, it will pick up at the step (rule in snakemake term) where it stopped last time. It will do nothing if it succeeded last time. The `--forceall` option can be used to enforce the re-run.
### DRAMv compatibility
点这里查看DRAMv工具的使用:[DRAM(Distilling and Refining Annotations of Metabolism,提取和精练代谢注释)工具安装和使用-CSDN博客文章浏览阅读251次。默认使用conda安装吧,也建议使用conda,pip安装其实都差不多,但容易破坏当前环境。所以其实这里还需要安装virsorter软件,virsorter的操作使用后面再补上。基于virsorter的结果进行注释,相当于过滤了。看不懂的百度翻译吧,反正需求内存不小。基因组完整注释和提取。https://blog.csdn.net/zrc\_xiaoguo/article/details/134578766?spm=1001.2014.3001.5502](https://blog.csdn.net/zrc_xiaoguo/article/details/134578766?spm=1001.2014.3001.5502 "DRAM(Distilling and Refining Annotations of Metabolism,提取和精练代谢注释)工具安装和使用-CSDN博客")
[DRAMv]( ) is a tool for annotating viral contigs identified by VirSorter. It needs two input files from VirSorter: 1) viral contigs, 2) `affi-contigs.tab` that have info on viral/nonviral and hallmark genes along contigs. In VirSorter2, these files can be generated by `--prep-for-dramv` flag.
rm -rf test.out
virsorter run --prep-for-dramv -w test.out -i test.fa -j 4 all
ls test.out/for-dramv
## Detailed description on output files
* final-viral-combined.fa
>
> identified viral sequences, including two types:
>
>
>
> + full sequences identified as viral (identified with suffix `||full`);
> + partial sequences identified as viral (identified with suffix `||{i}_partial`); here `{i}` can be numbers starting from 0 to max number of viral fragments found in that contig;
> + short (less than two genes) sequences with hallmark genes identified as viral (identified with suffix `||lt2gene`);
>
* final-viral-score.tsv
>
> This table can be used for further screening of results. It includes the following columns:
>
>
>
> + sequence name
> + score of each viral sequences across groups (multiple columns)
> + max score across groups
> + max score group
> + contig length
> + hallmark gene count
> + viral gene %
> + nonviral gene %
>
---
**NOTE**
Note that classifiers of different viral groups are not exclusive from each other, and may have overlap in their target viral sequence space, which means this information should not be used or considered as reliable taxonomic classification. We limit the purpose of VirSorter2 to viral idenfication only.
---
* final-viral-boundary.tsv
>
> only some of the columns in this file might be useful:
>
>
>
> + seqname: original sequence name
> + trim\_orf\_index\_start, trim\_orf\_index\_end: start and end ORF index on orignal sequence of identified viral sequence
> + trim\_bp\_start, trim\_bp\_end: start and end position on orignal sequence of identified viral sequence
> + trim\_pr: score of final trimmed viral sequence
> + partial: full sequence as viral or partial sequence as viral; this is defined when a full sequence has score > score cutoff, it is full (0), or else any viral sequence extracted within it is partial (1)
> + pr\_full: score of the original sequence
> + hallmark\_cnt: hallmark gene count
> + group: the classifier of viral group that gives high score; this should **NOT** be used as reliable classification
>
---
**NOTE**
VirSorter2 tends to sometimes overestimate the size of viral sequence during provirus extraction procedure in order to achieve better sensitity. We recommend cleaning these provirus predictions to remove potential host genes on the edge of the predicted viral region, e.g. using a tool like CheckV ([Bitbucket]( )).
---
## Training customized classifiers
VirSorter2 currently has classifiers of five viral groups (dsDNAphage, NCLDV, RNA, ssNA virus, and *lavidaviridae*). It's designed for easy addition of more classifiers. The information of classifiers are store in the database (`-d`) specified during [setup step]( ). For each viral group, it needs four files below:
为了做好运维面试路上的助攻手,特整理了上百道 **【运维技术栈面试题集锦】** ,让你面试不慌心不跳,高薪offer怀里抱!
这次整理的面试题,**小到shell、MySQL,大到K8s等云原生技术栈,不仅适合运维新人入行面试需要,还适用于想提升进阶跳槽加薪的运维朋友。**

本份面试集锦涵盖了
* **174 道运维工程师面试题**
* **128道k8s面试题**
* **108道shell脚本面试题**
* **200道Linux面试题**
* **51道docker面试题**
* **35道Jenkis面试题**
* **78道MongoDB面试题**
* **17道ansible面试题**
* **60道dubbo面试题**
* **53道kafka面试**
* **18道mysql面试题**
* **40道nginx面试题**
* **77道redis面试题**
* **28道zookeeper**
**总计 1000+ 道面试题, 内容 又全含金量又高**
* **174道运维工程师面试题**
> 1、什么是运维?
> 2、在工作中,运维人员经常需要跟运营人员打交道,请问运营人员是做什么工作的?
> 3、现在给你三百台服务器,你怎么对他们进行管理?
> 4、简述raid0 raid1raid5二种工作模式的工作原理及特点
> 5、LVS、Nginx、HAproxy有什么区别?工作中你怎么选择?
> 6、Squid、Varinsh和Nginx有什么区别,工作中你怎么选择?
> 7、Tomcat和Resin有什么区别,工作中你怎么选择?
> 8、什么是中间件?什么是jdk?
> 9、讲述一下Tomcat8005、8009、8080三个端口的含义?
> 10、什么叫CDN?
> 11、什么叫网站灰度发布?
> 12、简述DNS进行域名解析的过程?
> 13、RabbitMQ是什么东西?
> 14、讲一下Keepalived的工作原理?
> 15、讲述一下LVS三种模式的工作过程?
> 16、mysql的innodb如何定位锁问题,mysql如何减少主从复制延迟?
> 17、如何重置mysql root密码?
**网上学习资料一大堆,但如果学到的知识不成体系,遇到问题时只是浅尝辄止,不再深入研究,那么很难做到真正的技术提升。**
**需要这份系统化的资料的朋友,可以添加V获取:vip1024b (备注运维)**

**一个人可以走的很快,但一群人才能走的更远!不论你是正从事IT行业的老鸟或是对IT行业感兴趣的新人,都欢迎加入我们的的圈子(技术交流、学习资源、职场吐槽、大厂内推、面试辅导),让我们一起学习成长!**
MQ是什么东西?
> 14、讲一下Keepalived的工作原理?
> 15、讲述一下LVS三种模式的工作过程?
> 16、mysql的innodb如何定位锁问题,mysql如何减少主从复制延迟?
> 17、如何重置mysql root密码?
**网上学习资料一大堆,但如果学到的知识不成体系,遇到问题时只是浅尝辄止,不再深入研究,那么很难做到真正的技术提升。**
**需要这份系统化的资料的朋友,可以添加V获取:vip1024b (备注运维)**
[外链图片转存中...(img-p7OhxUCq-1713414540467)]
**一个人可以走的很快,但一群人才能走的更远!不论你是正从事IT行业的老鸟或是对IT行业感兴趣的新人,都欢迎加入我们的的圈子(技术交流、学习资源、职场吐槽、大厂内推、面试辅导),让我们一起学习成长!**

















 5860
5860

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









