






首先打开GaussView画一个水分子,
【之后Calculate下setup,设置一系列后retain,在之后save】具体见之前操作
之后用Gaussian打开,整体设置情况见下图
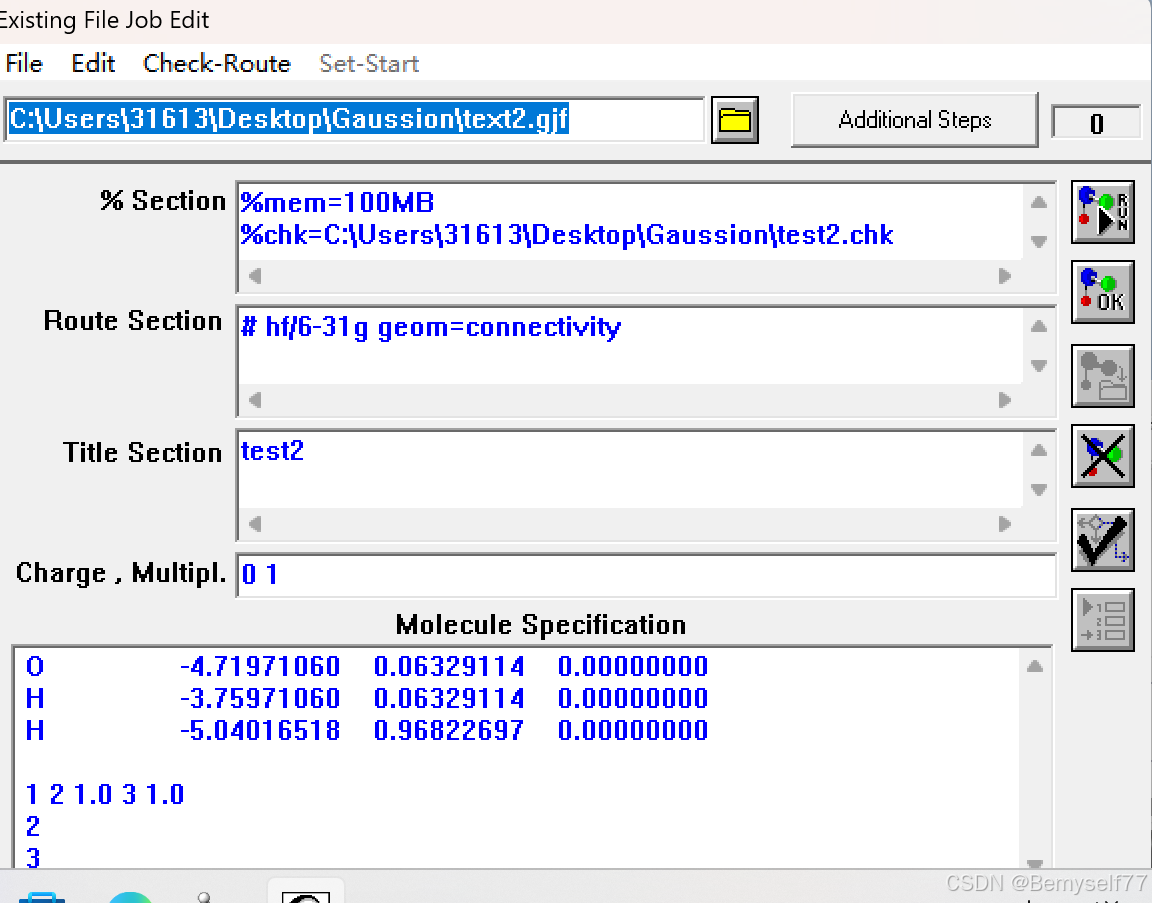
点击运行
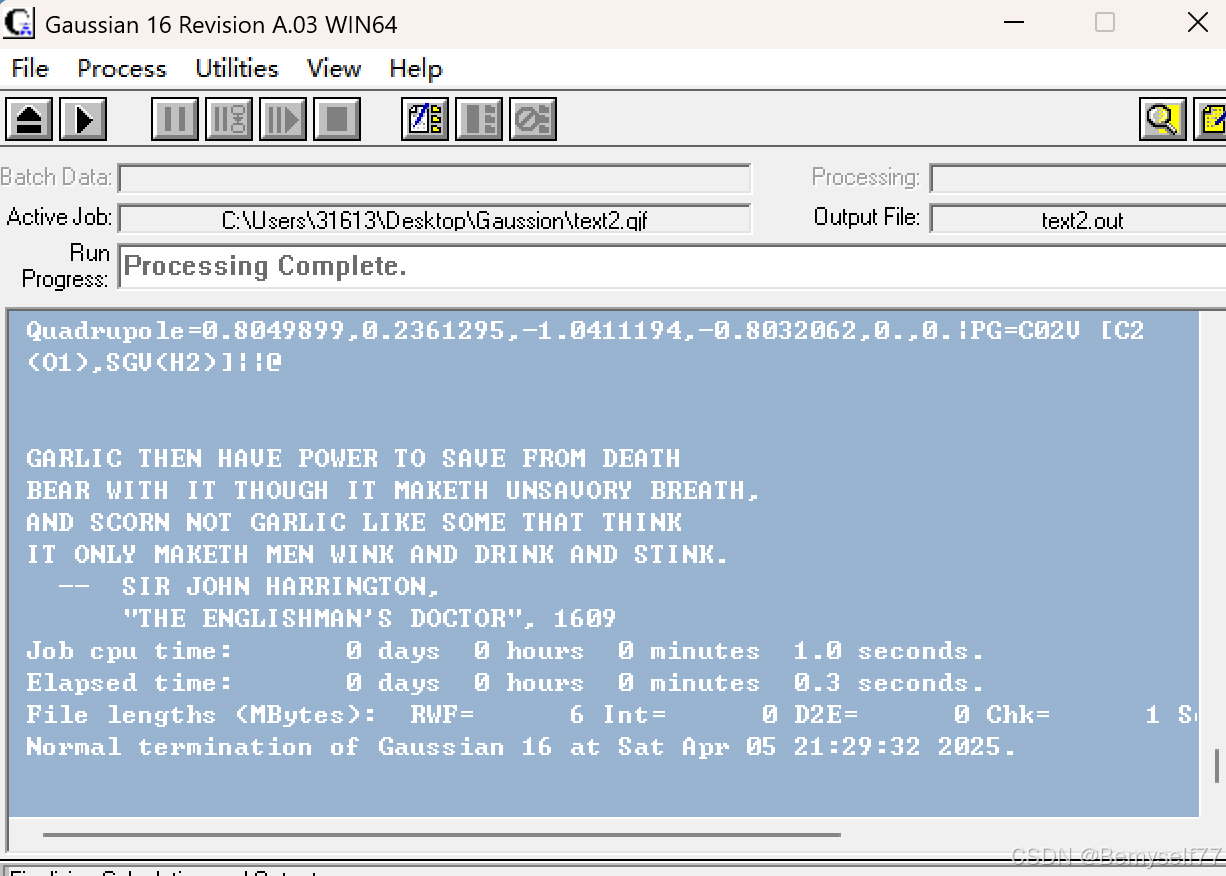
出现Processing Complete说明已经计算完了。点击![]() 进行打开
进行打开
打开后内容如下
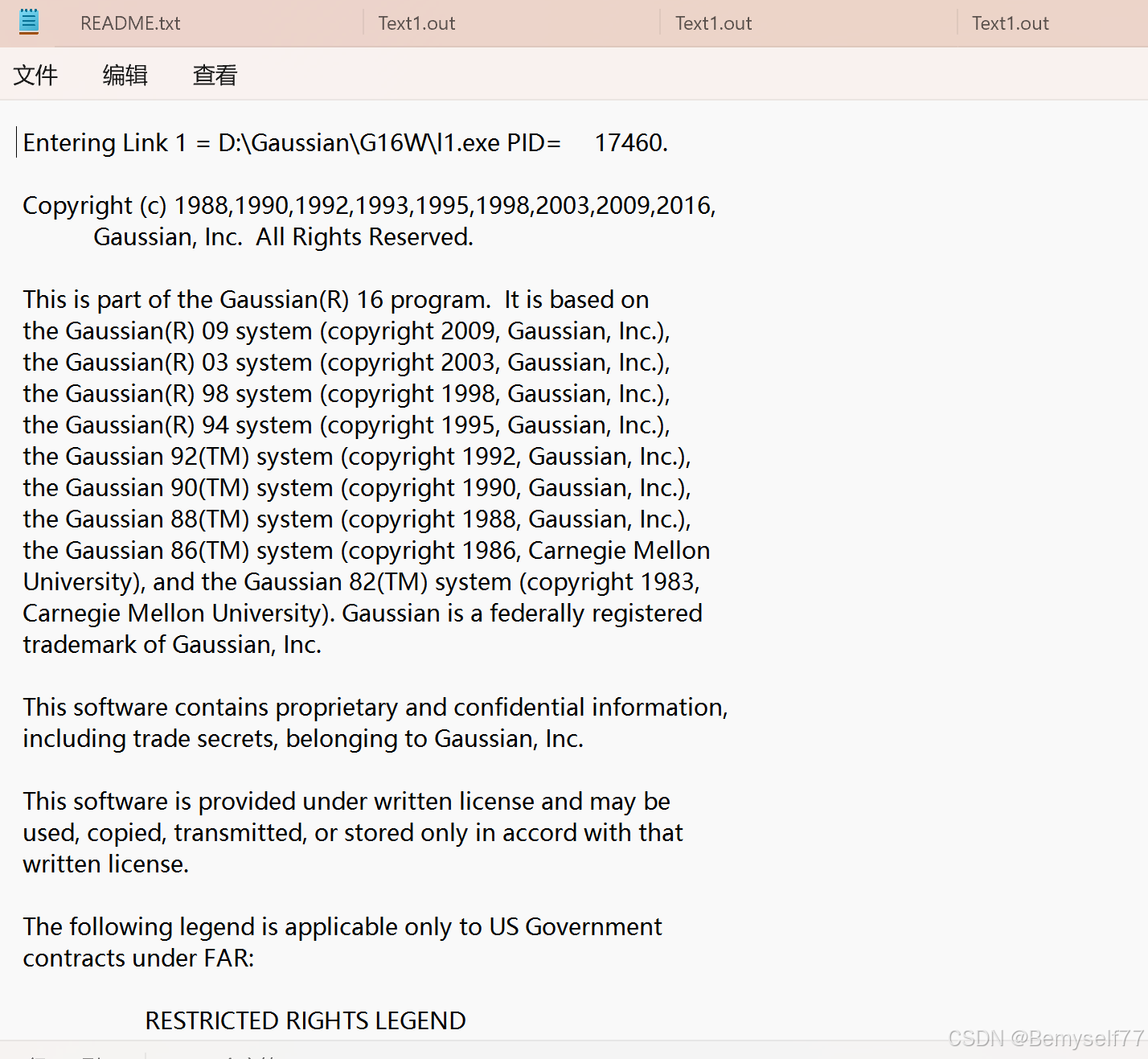
这样的一堆就是通过Gaussian计算得到的,也就是说可以将Gaussian理解为一个模块化的程序,他由很多不同的模块组成,不同模块有不同的功能,可以通过模块来实现数据的计算,这个模块就是link,我们之前在设置中设置了 link0,可以把他理解为 具有初始化程序的控制模块。
整个输出首先是文件和程序的定位,接着往下走是有关于版权的引用

上图下方是使用的软件的版本的信息:Gaussian 16

之后看的出来是将整个输入文件的内容进行了复制,chk文件名,计算路径,非标准执行路径,计算标题名称,电荷自旋多重度,以及坐标。
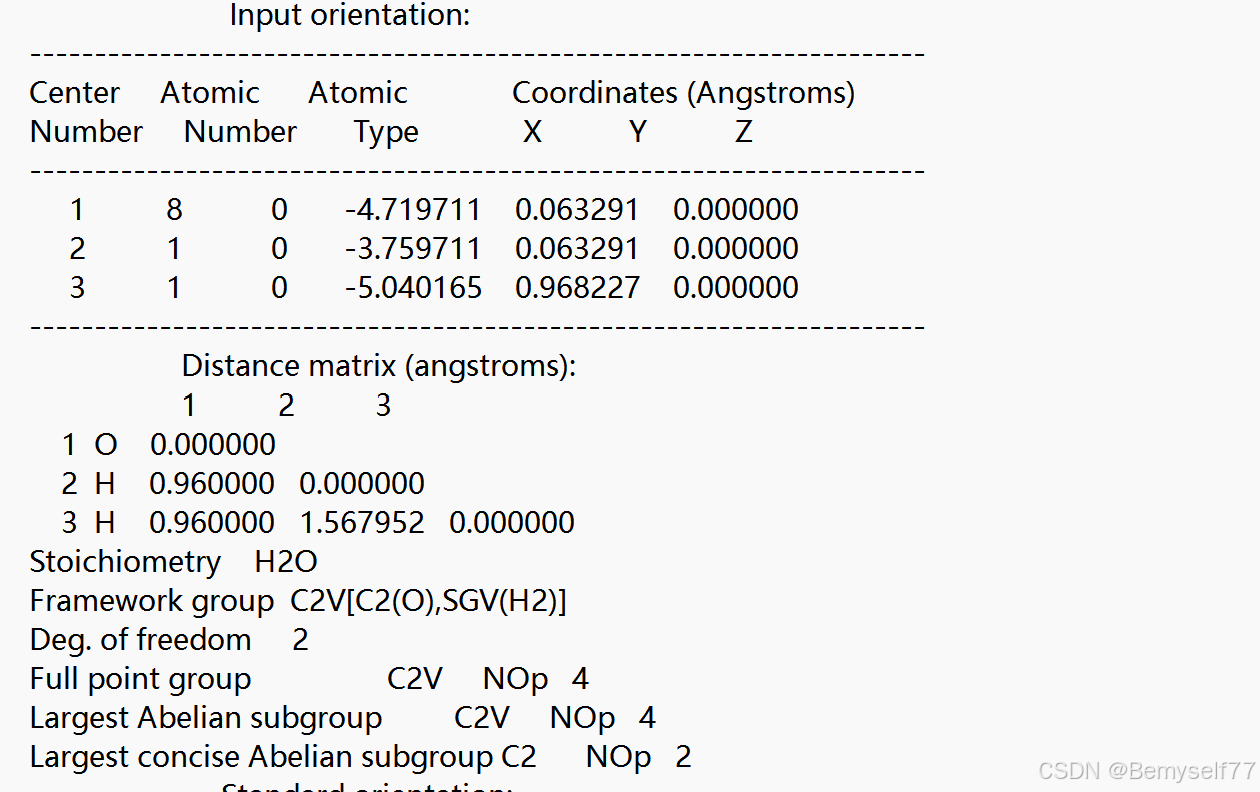
这一部分记录了 Input orientation(输入取向)部分,原子核间距以及键长
之后是对称性,通过程序判定分子所属点群

Standard orientation,是程序在计算时,其内部会采用的坐标体系,可用于优化性能,坐标体系的原点是分子的核电荷中心。

standard basis是基组的信息,之后再往下

Intitial guess为初试猜测区, SCF Done为通过单点能计算所得到的能量,注意能量单位是Hartree,1Hartree=2625.5 kj/mol;conv表示自洽场计算,“conv” 用于描述电子密度和能量的迭代计算是否达到收敛标准【自洽场计算是通过不断迭代调整分子轨道和电子密度,直到前后两次迭代的能量变化和轨道系数变化小于某个预设的阈值(收敛标准),此时认为计算达到收敛。】,D-08是10的负8次方。-V/T是维里系数,其中 V 代表势能(Potential Energy),T 代表动能(Kinetic Energy),该数字约等于2,则表明这个计算没有问题
接下来就是布局分析,首先可以看到分子的基态光谱
“Condensed to atoms (all electrons):” 这部分内容 表示的是将电子密度等相关性质按照原子进行凝聚(即把总的电子相关性质分配到各个原子上 )。

Muliken charges指被估算分子中每一个原子上的总电荷,可以看到氧原子带正电荷,氢原子带负电荷,且整体呈中性
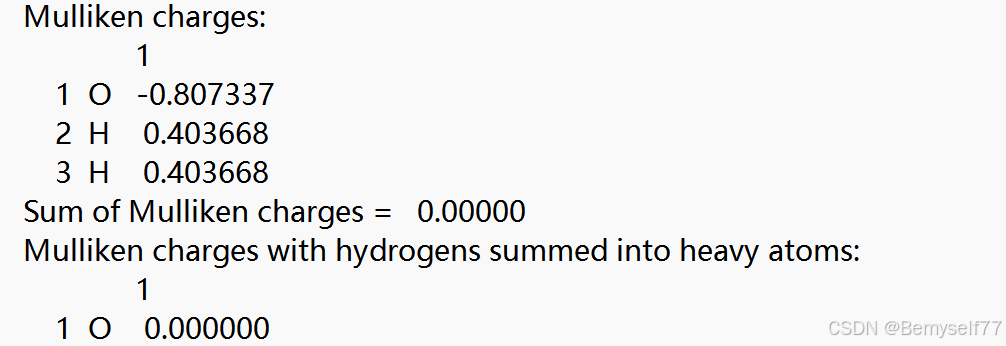
接下来是偶极矩和多极矩,不同于之前的 库仑・米(C・m),单位是德拜(Debye,D),故要进行换算,1D≈3.33564×10−30C⋅m,对于多极矩只有当偶极矩为0时多极矩才算是有用的

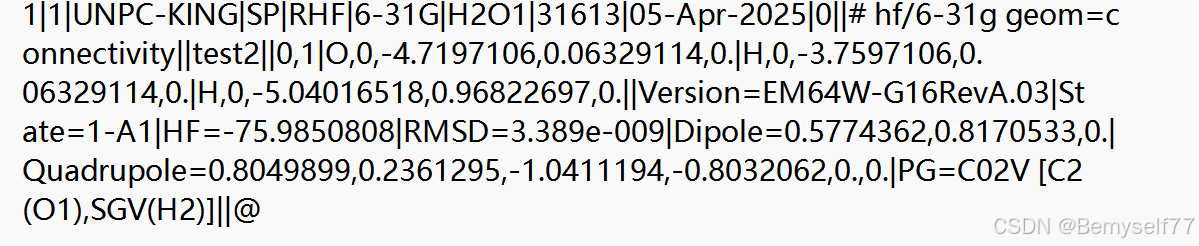
接下来是对整块计算的总结不同的内容使用|进行隔开,在这里可以看到光谱项State,总能量HF等
{HF = - 75.9850808:这里的 “HF” 表示 Hartree - Fock 能量,即通过 Hartree - Fock 方法计算得到的体系能量;RMSD = 3.389e - 009:“RMSD” 是均方根偏差(Root - Mean - Square Deviation),在结构优化等计算中,它用于衡量原子坐标的变化程度或收敛情况。数值越小,说明计算结果越稳定,原子坐标的变化越小。Dipole = 0.5774362,0.8170533,0.0:表示分子的偶极矩,通过这些分量可以了解分子在不同方向上的极性特征。Quadrupole = 0.8049899,0.2361295, - 1.0411194, - 0.8032062,0.,0.:表示分子的四极矩,四极矩描述了分子电荷分布的非球形特征,这些数值反映了分子在不同方向上的电荷分布情况。PG = C02V [C2(O1),SGV(H2)]:“PG” 代表点群(Point Group),“C02V” 表示分子的点群对称性,中括号内的内容进一步说明了这种对称性在原子上的体现,类似于之前对框架群的描述,反映了分子的对称特征。}
最后是计算时间和文件大小,最后出现Normal termination出现后就表示计算顺利的结束了
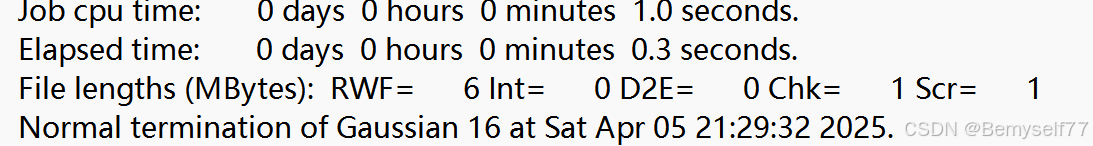

















 1026
1026

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









