






1写在前面
之前我们在质控(QC)篇介绍了Normalization的重要性。😘
scRNAseq是一个高维度的数据,相对比较复杂,不同细胞,不同平台,相差较多,所以library大小差异较大,我们需要做一个Normalization,常用方法包括:👇
UQ, SF, CPM, RPKM, FPKM, TPM。🤒
Note! 如果你使用的是Cufflinks或者RSEM进行定量。
恭喜你, 不需要normalization,因为这类方法已经考虑到的library大小不同的问题了。
2用到的包
rm(list = ls())
library(tidyverse)
library(SingleCellExperiment)
library(scater)
library(scran)
library(scRNA.seq.funcs)
3示例数据
这里我们用一下之前介绍的counts文件和annotation文件,然后通过SingleCellExperiment创建SingleCellExperiment格式的文件,并且经过初步过滤,ID转换等。
load("umi_umiqc.Rdata")
umi.qc
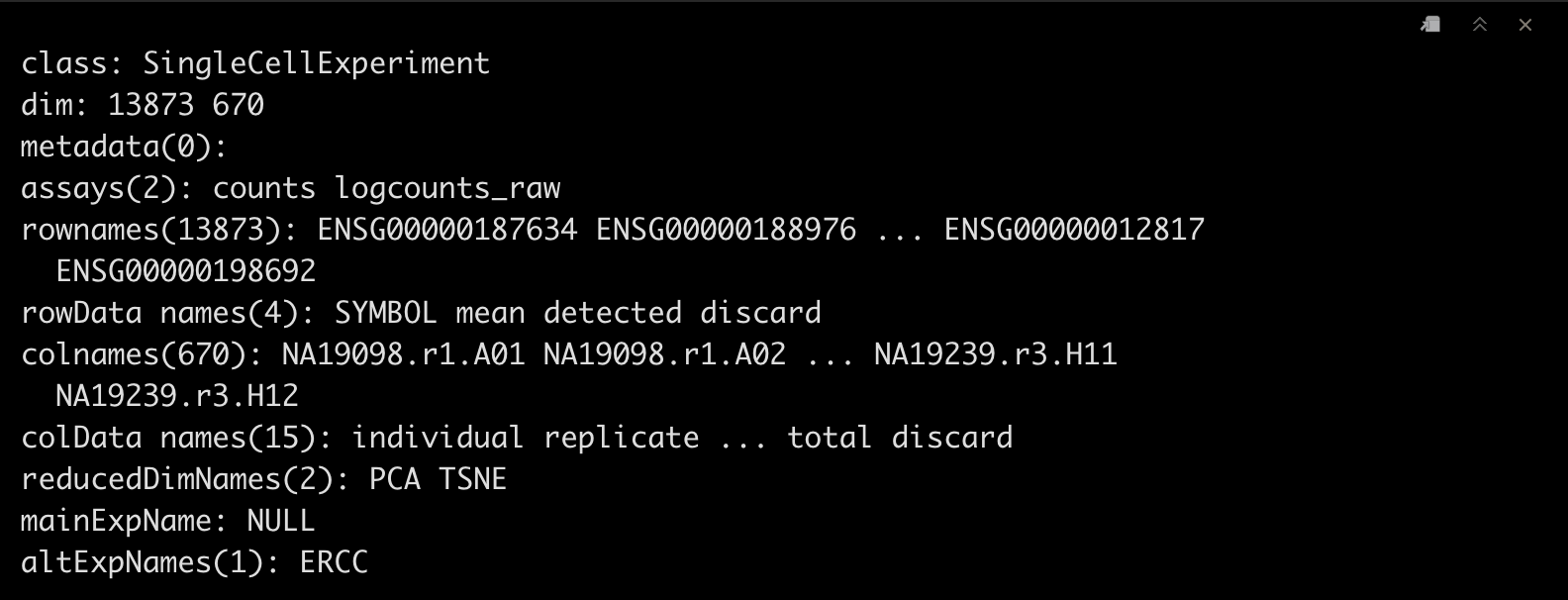
4PCA-rawdata
这里我们用取过log后的raw_counts进行PCA绘图。
umi.qc <- runPCA(umi.qc, exprs_values = "logcounts_raw")
plotPCA(umi.qc, colour_by = "batch", size_by = "detected", shape_by = "individual")
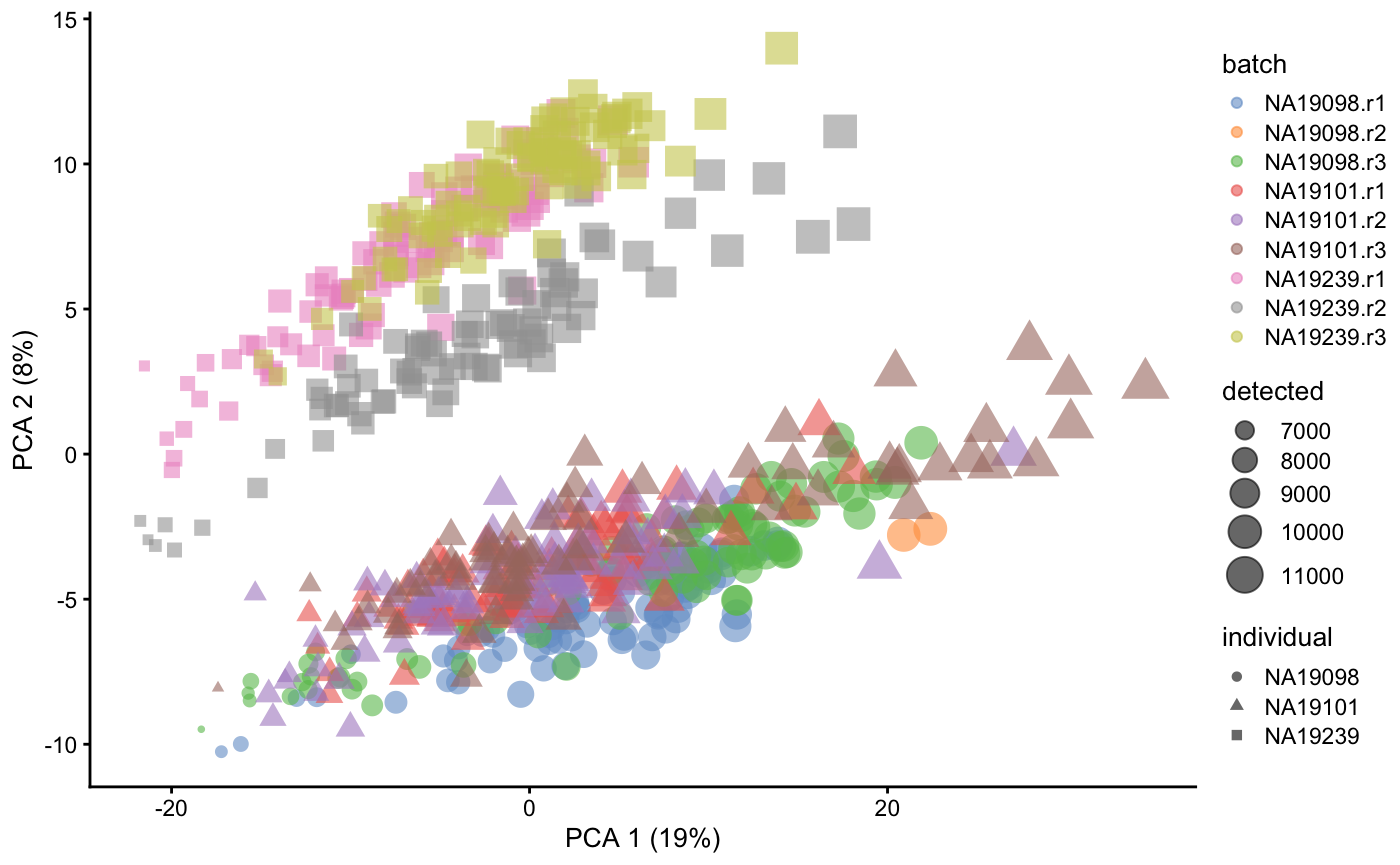
5PCA-CPM
这里我们先做一个CPM,然后取log,再做一下PCA。 聚类明显要好一些了!😂
logcounts(umi.qc) <- log2(calculateCPM(umi.qc) + 1)
umi.qc <- runPCA(umi.qc)
plotPCA(umi.qc, colour_by = "batch", size_by = "detected", shape_by = "individual")
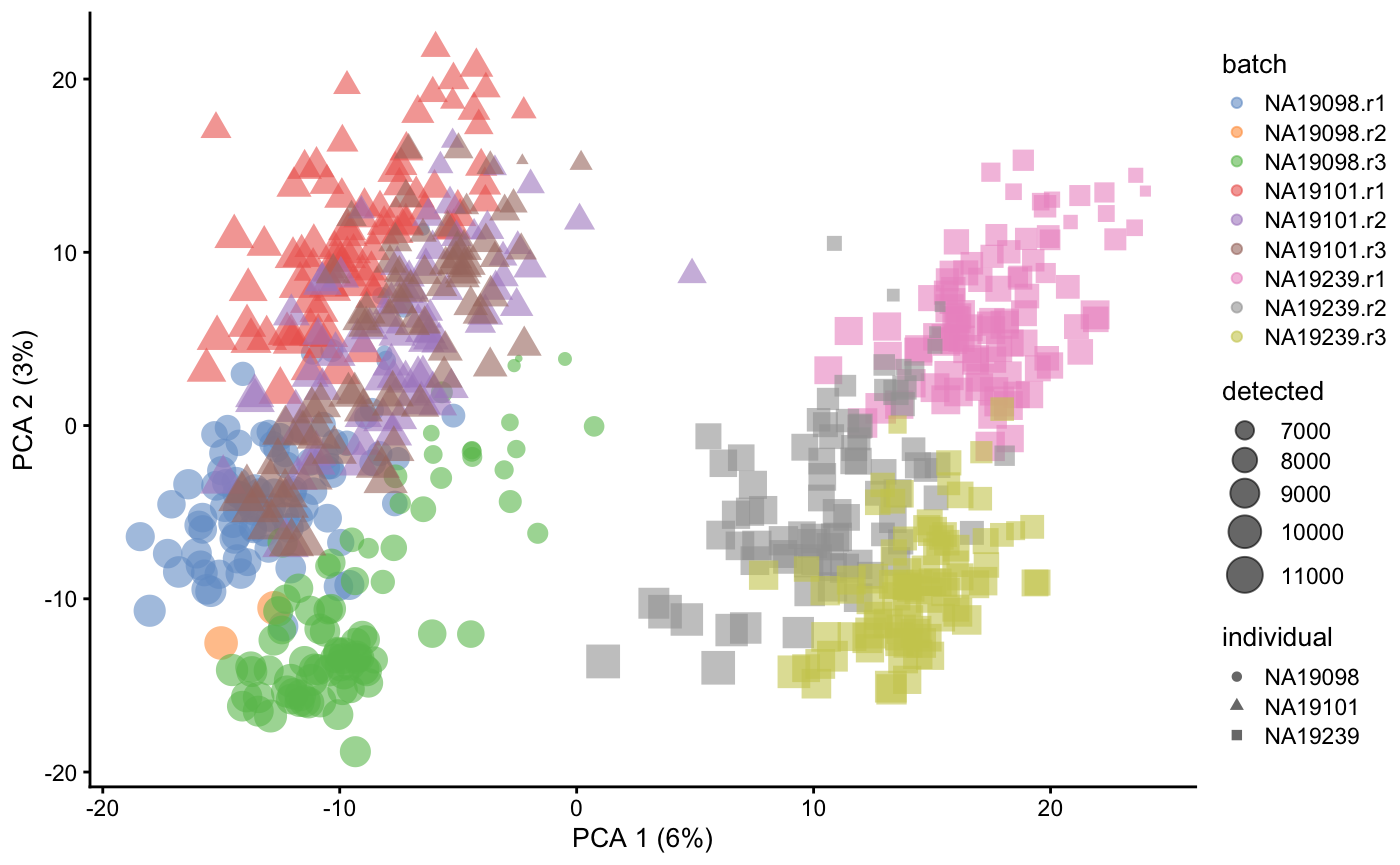
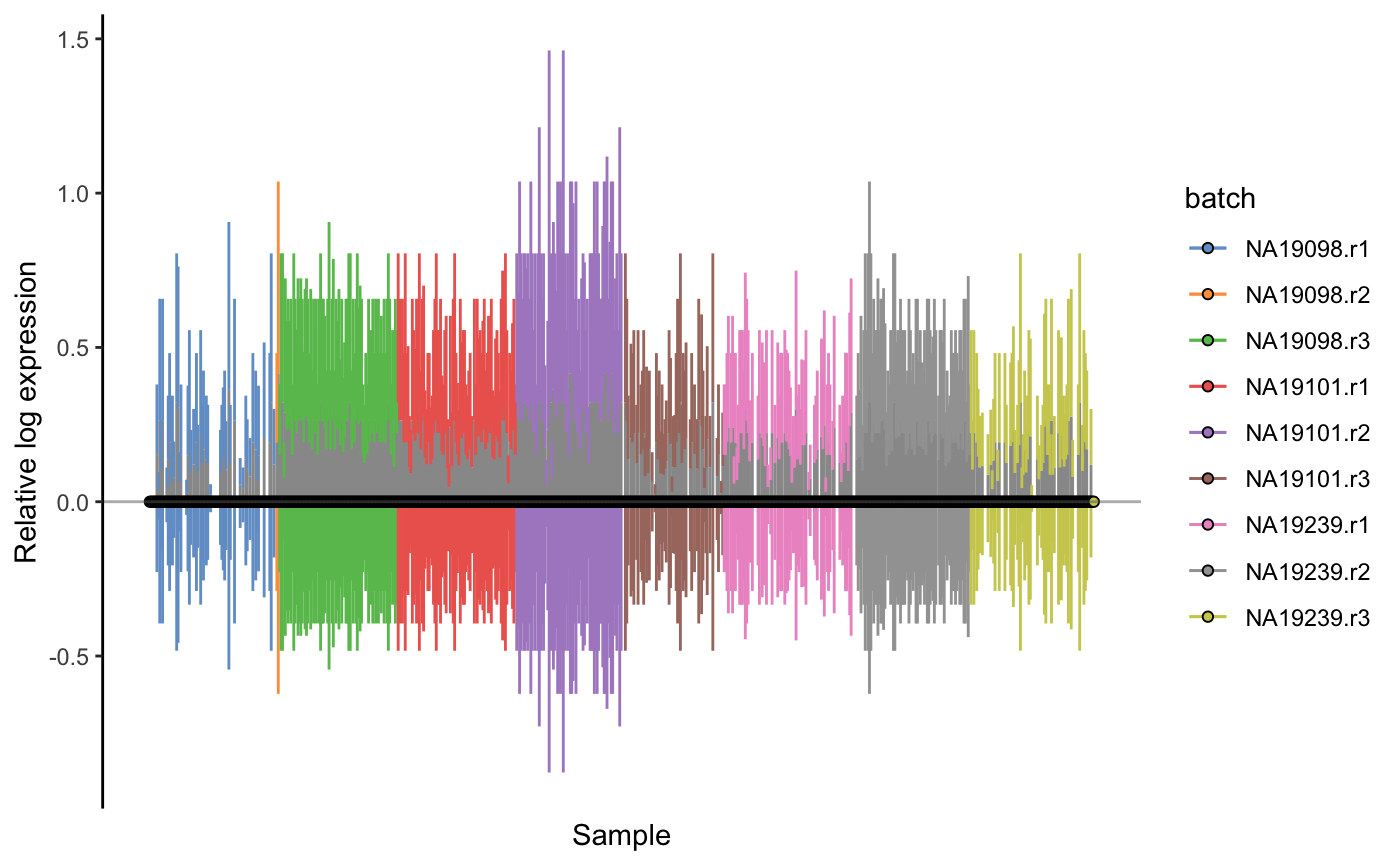
6RLE-rawdata
这里我们引入一个新的可视化方式,叫relative log expression (RLE) plots。
先对raw_counts做一个RLE plots吧, 这里可以看出有明显批次效应。
plotRLE(umi.qc, exprs_values = "logcounts_raw",colour_by = "batch") + ggtitle("RLE plot for logcounts_raw")
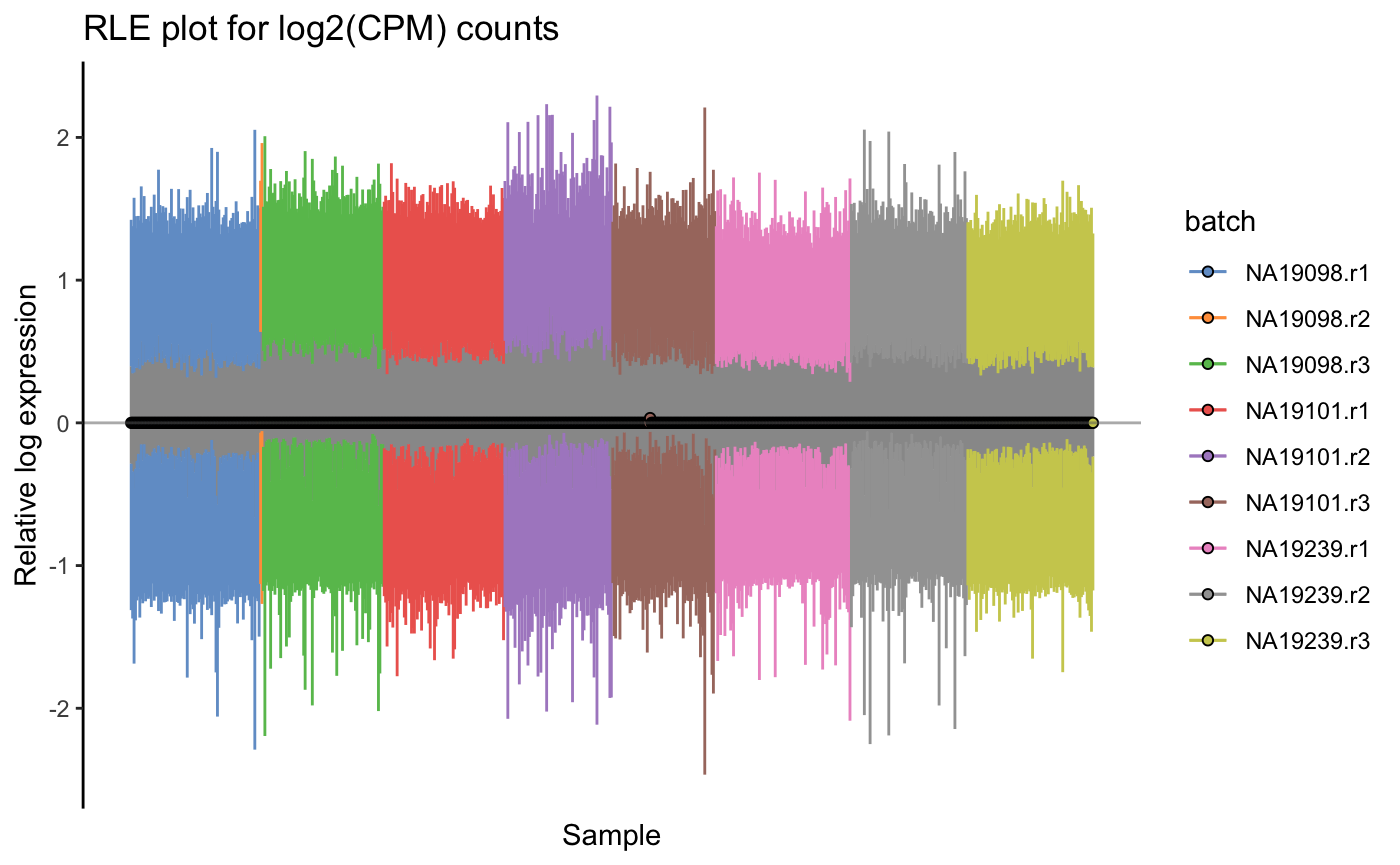
7RLE-CPM
再看一下CPM后的结果吧,非常明显!🥳
plotRLE(umi.qc, exprs_values = "logcounts",colour_by = "batch") + ggtitle("RLE plot for log2(CPM) counts")
8其他方法
基于CPM的Normalization方法,或者其他类似的方法,有一个前提,就是假设所有细胞含有相似数量的RNA,但这有时候是不对的。🤒
这里我们介绍一下scran包,通过反卷积的方法进行Normalization。🤗
8.1 聚类
qclust <- quickCluster(umi.qc, min.size = 30)
table(qclust)

8.2 计算sizefactor
接着我们来计算一下sizeFactor,用于后面的Normalization。🧐
umi.qc <- computeSumFactors(umi.qc, clusters = qclust)
8.3 正式Normalization
开始log和Normalization。🤩
umi.qc <- logNormCounts(umi.qc)
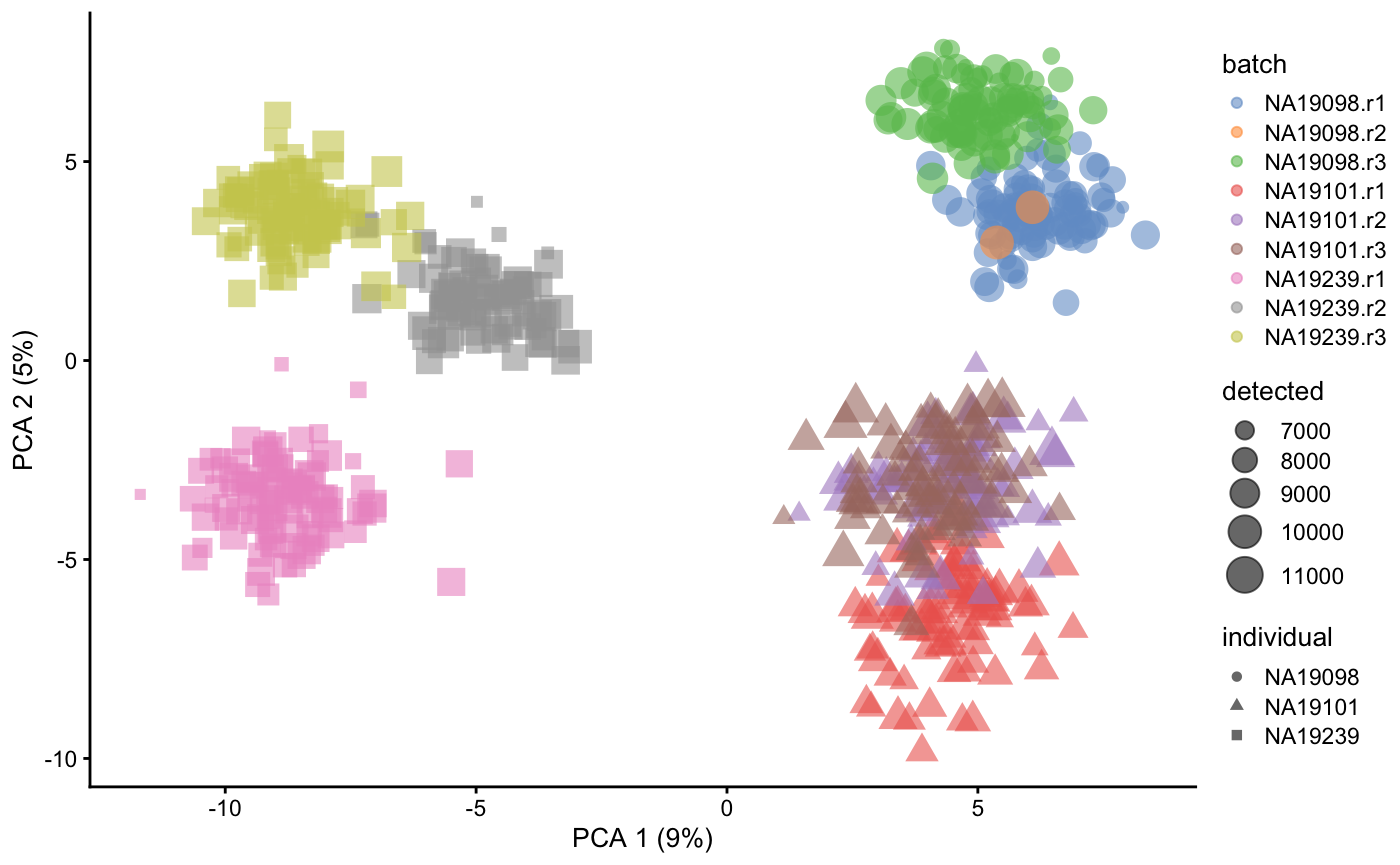
8.4 可视化-PCA
umi.qc <- runPCA(umi.qc)
plotPCA(umi.qc, colour_by = "batch",size_by = "detected", shape_by = "individual")
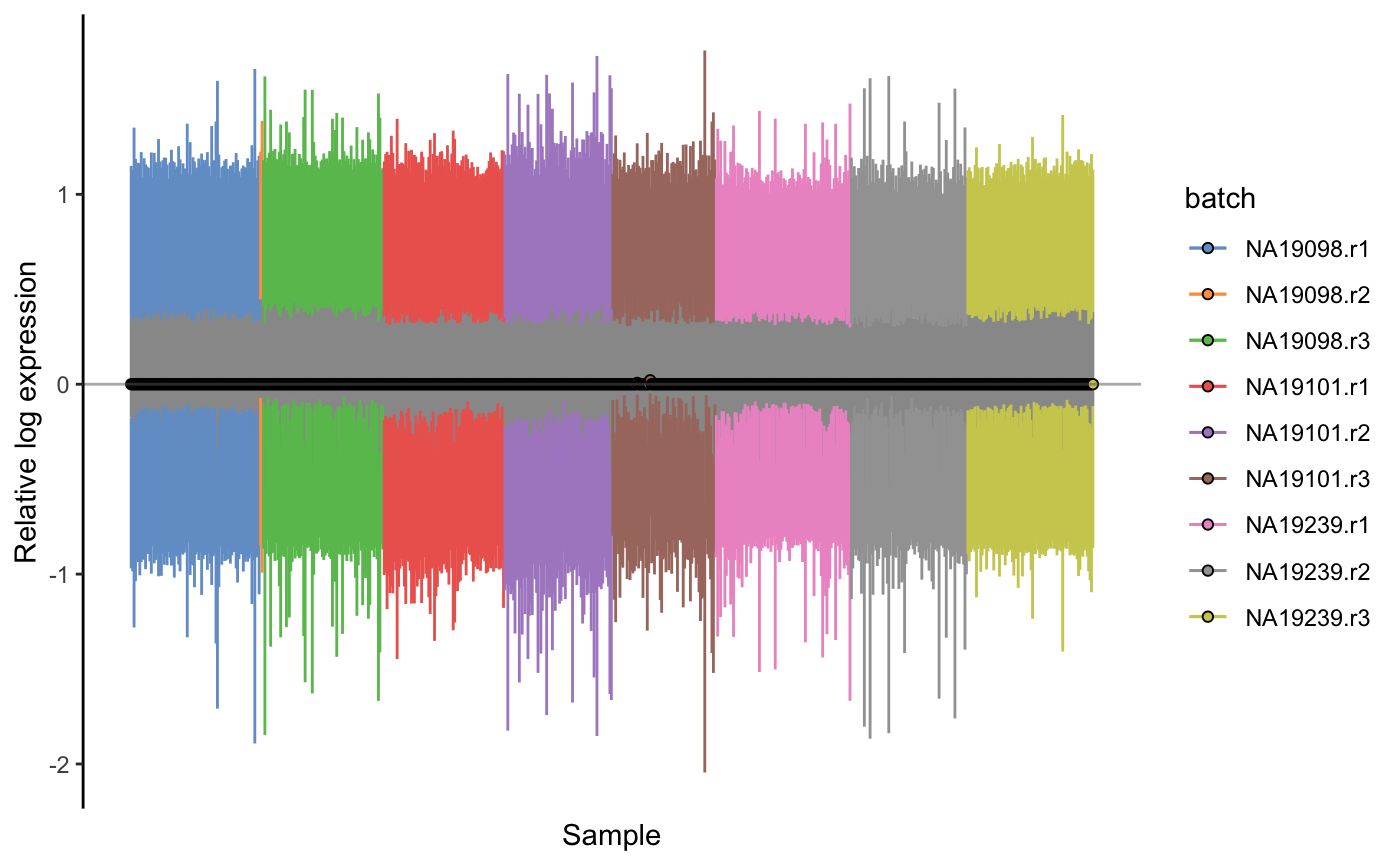
8.5 RLE plot
plotRLE(umi.qc, exprs_values = "logcounts",colour_by = "batch")
8.6 补充一下
如果你已经做过Normalization,再做一次的话,这个size factors会是负数,严重影响你的结果。🫠
我们可以check一下。✌️
summary(sizeFactors(umi.qc))

9downsampled data
这里再介绍一个scRNA.seq.funcs包的Down_Sample_Matrix函数。
采用了downsample,降采样的方法,具体概念不介绍了,大家自行google。
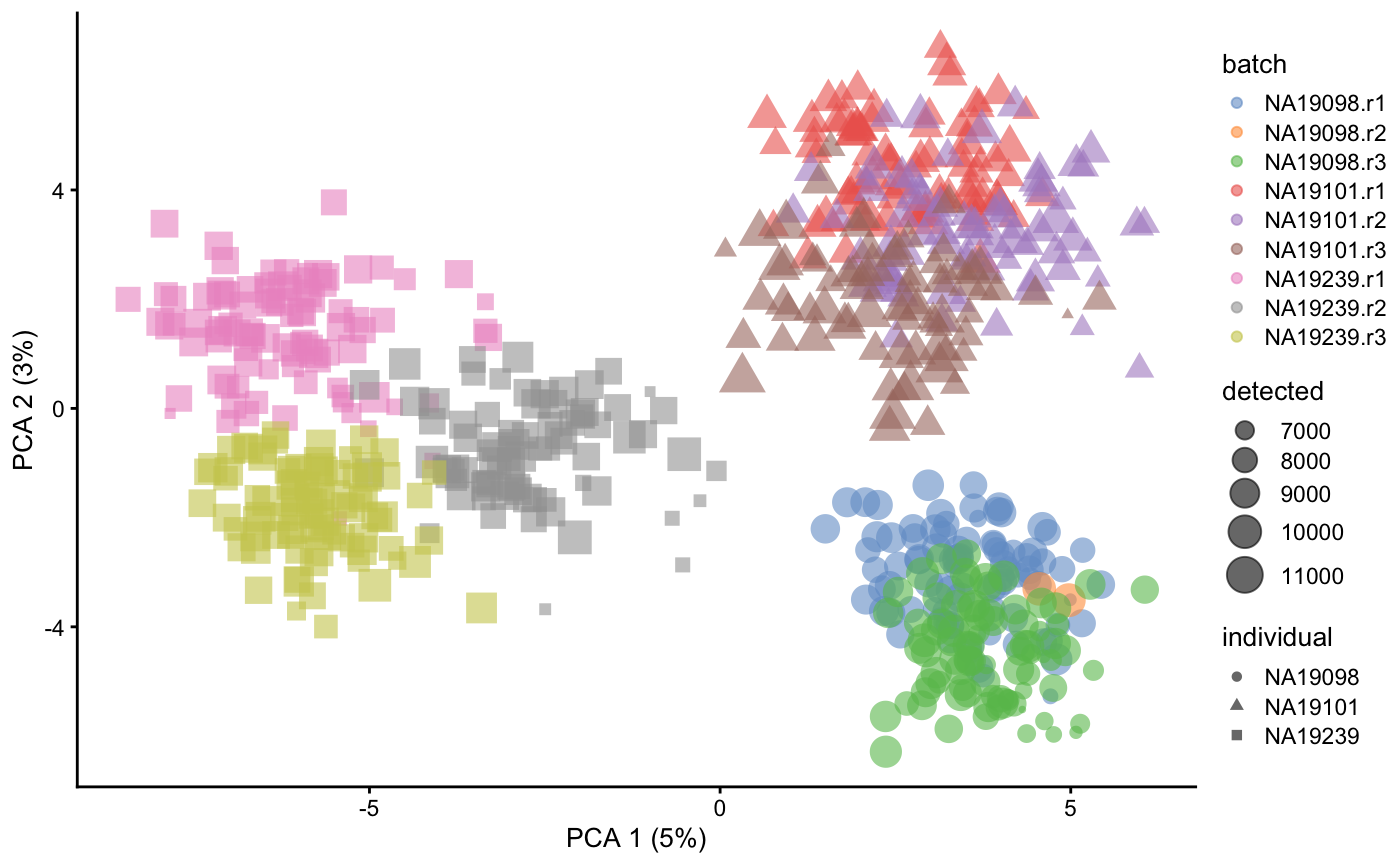
9.1 PCA
logcounts(umi.qc) <- log2(Down_Sample_Matrix(counts(umi.qc)) + 1)
umi.qc <- runPCA(umi.qc)
plotPCA(umi.qc,colour_by = "batch",size_by = "detected", shape_by = "individual")
9.2 RLE plot
plotRLE(umi.qc, exprs_values = "logcounts",colour_by = "batch")

需要示例数据的小伙伴,在公众号回复
scRNAseq获取吧!点个在看吧各位~ ✐.ɴɪᴄᴇ ᴅᴀʏ 〰
本文由 mdnice 多平台发布















 5149
5149











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









