







目录
2.8 最后导出模块中 TOM值 的数据,导入 cytoscape 作图
2.9 用 cytoscape 将上述特定模块中的基因以互作网络图形式展现,以一定的标准来选择 hub gene 作为后续研究的重点
一. WGCNA 安装及介绍
1.1 安装及出现问题的解决方案
- WGCNA 官网安装地址(国外):https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/
WGCNA 安装前提:需要安装 R 和 Rstudio
WGCNA 安装命令(在 Rstudio 中输入命令,从 CRAN 自动安装,比手动方便):- install.packages(“ BiocManager”)
BiocManager :: install(“ WGCNA”)
出现报错的解决方案:- 查过手动安装未加载的包,失败,手动易出现更多问题
最终成功的方案:重复执行 CRAN 安装方法的命令1.2 WGCNA 概念
- 参考文档:https://mp.weixin.qq.com/s/n2DDYAvnnDO5Gw8QsrXCXQ?
WGCNA 分析:加权基因 共表达网络分析- 该分析方法旨在:
- 寻找协同表达的基因模块(module)
- 探索基因网络与关注的表型之间的关联关系,以及网络中的核心基因
- 应用场景及研究方向:不同器官或组织类型发育调控、同一组织不同发育调控、非生物胁迫不同时间点应答、病原菌侵染后不同时间点应答
基本思路:
- 在大样本(推荐5组 / 15个样品以上)基因表达数据中,找出具有 相似表达谱 的基因,归于同一模块(module)
- 对模块进行区分,通过:模块特征值(module eigengene)/ 枢纽基因(hub gene)
- 计算 模块与模块相关性、模块与样本性状相关性
- 筛选 与性状高度相关 的模块,分析此模块内部的基因,找到所需的目标基因
同类方法比较:- 与普通聚类方法的不同:将基因表达值的相关系数取了 n次幂,使相关系数分布更加合理
- 与其他共表达网络分析的不同:加入了软阈值、权重网络的概念
WGCNA 分析(属于预测范畴)步骤:
- 数据输入和数据清洗
- 建设表达网络和模块检测
- 筛选与表型相关的模块
- 使用 WGCNA 进行网络可视化
WGCNA 分析(属于预测范畴)步骤(详细版):
- 计算任意基因之间的相关系数 —— 为了衡量两个基因是否具有相似表达模式,需要设置 阈值 筛选
- 高于阈值就是相似的,但如果将阈值设为 0.8,就很难说明 0.8 和 0.79 两个是有显著差别的
- WGCNA分析时采用:相关系数加权值(对基因相关系数取N次幂),使得网络中的基因之间的连接服从 无尺度网络分布 (scale-freenetworks),这种算法更具生物学意义
- 基于基因相关系数,构建分层聚类树,聚类树的不同分支、不同颜色代表不同的基因模块
- 基于基因的加权相关系数,将基因按照表达模式进行分类,将模式相似的基因归为一个模块
- 几万个基因通过基因表达模式,被分成了几十个模块,是一个 提取归纳信息 的过程
- 得到模块之后的分析: 模块的功能富集、模块与性状的 相关性、模块与样本的 相关系数
- 挖掘模块的关键信息:找到模块的核心基因、利用关系预测基因功能
二. WGCNA 实例及总结
- 此节包含了 —— 表达谱文件 + 样本性状文件,总结了 WGCNA 使用流程
2.1 导入表达谱数据 + 样品性状数据
- Mine:
- 表格内容:每个基因在不同组织当中的表达值
- 表头是样本信息(比如花生哪个组织采样,叶子、花等等),第一列是花生基因ID
Example:- 表达谱矩阵可看出:有 3600个基因、136个样本;
- 样品性状矩阵可看出:有 361个样本、7个性状;
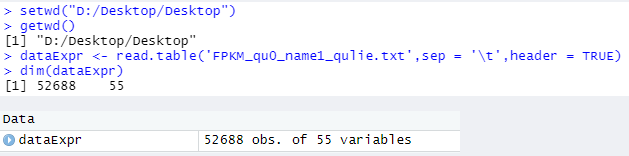
———————————————————————————————————————————————————————————————————— Mine: ———————————————————————————————————————————————————————————————————— // 加载 WGCNA软件 library(WGCNA) options(stringsAsFactors = FALSE) // 允许R语言程序最大线程运行 allowWGCNAThreads() // 调换文件打开根目录 > setwd("D:/Desktop/Desktop/大创") // 检查当前根目录 > getwd() [1] "D:/Desktop/Desktop" // 读取 txt 数据文件,并赋值给 dataExpr变量 > dataExpr <- read.table('FPKM_qu0_name1_qulie.txt',sep = '\t',header = TRUE) // dim():查看变量维数 > dim(dataExpr) // 52688 个基因,55个样本 [1] 52688 55 ———————————————————————————————————————————————————————————————————— Example: ———————————————————————————————————————————————————————————————————— dataExpr <- read.csv(file = "Sample_Expr.csv") // 表达谱文件 dim(dataExpr) // 表达谱矩阵 [1] 3600 136 dataTraits <- read.csv(file = "ClinicalTraits.csv") // 样本性状文件 dim(dataTraits) // 样品性状矩阵 [1] 361 7
2.2 数据预处理
- 通过过滤低表达量、低变异系数的基因,以减少参与后续分析的基因数目,提高结果可靠性
- 由于实验数据的特殊性,就不做前一步处理了
- 将矩阵写为符合 WGCNA 要求的形式:行名为 gene,列名为样品
- 让性状数据和表达谱数据保持一致(一一对应)
// as.data.frame():将已存在的数据转为数据框格式; // t():将行列数据转置 dataExpr <- as.data.frame(t(dataExpr)) // colnames():修改矩阵的列名 colnames(dataExpr) <- dataExpr[1,] dataExpr <- dataExpr[-1,] // unlist():将list结构的数据,变成非list的数据 // as.numeric:转为数字格式 dataExpr <- unlist(apply(dataExpr, 1, as.numeric)) // $Mice:缺失值填补 // match(x,y):返回一个和 x 长度相同,同时和 y 中元素相等的向量 match_trait <- match(rownames(dataExpr), dataTraits$Mice) rownames(dataTraits) <- dataTraits$Mice // c(): 将括号中的元素连接起来,不创建向量,c(1, 2:4),结果为 1 2 3 4 // paste(): 连接括号中的元素,创建向量,paste(1, 2:4),结果为 “1 2” “1 3” “1 4” Traits <- dataTraits[match_trait, -c(1,2)]
2.3 筛选合适的阈值
- Mine:
- 选一个合适的值,使样本变为一个无尺度分布:
- 少部分基因处于绝对优势的位置(表达量高),大部分处于劣势(表达量低)
- 对基因间的相关系数取幂指数,最终确定合适的阈值
// 选择一组阈值 把 1-20 写成数组赋值给 powers // seq():seq(2,10,2),会生成一组数:2 4 6 8 10 powers <- c(c(1:10), seq(from = 12, to=20, by=2)) sft <- pickSoftThreshold(dataExpr, powerVector = powers) // 绘制结果图 par(mfrow = c(1, 2)) plot(sft$fitIndices[, 1], -sign(sft$fitIndices[, 3]) * sft$fitIndices[, 2], xlab = "Soft Threshold (power)", ylab = "SFT, signed R^2", type = "n", main = paste("Scale independence")) text(sft$fitIndices[, 1], -sign(sft$fitIndices[, 3]) * sft$fitIndices[, 2], labels = powers, col = "red") abline(h = 0.9, col = "red") plot(sft$fitIndices[, 1], sft$fitIndices[, 5], type = "n", xlab = "Soft Threshold (power)", ylab = "Mean Connectivity", main = paste("Mean connectivity")) text(sft$fitIndices[, 1], sft$fitIndices[, 5], labels = powers, col = "red")
- 根据上图中的红线位置,确定 power 阈值选为7,R² = 0.9
- WGCNA 建议的阈值,输入sft 查看,列表第一项就是估计的最优值
Mine:
2.4 分割模块,作网络图
- 分割模块:
- 采用动态剪切树算法(dynamic tree cutting),分为两种:
- 一步法:简单明了,一步出结果
- 分步法:细调参数,以达到满意结果
- 官网没说哪种适合什么条件,都可以用,参考如下:
- http://blog.sina.com.cn/s/blog_61f013b80101lcpr.html
net <- blockwiseModules(dataExpr, power = 7, maxBlockSize = 5000, TOMType = "unsigned", minModuleSize = 30, reassignThreshold = 0, mergeCutHeight = 0.25, numericLabels = TRUE, pamRespectsDendro = FALSE, saveTOMs = TRUE, saveTOMFileBase = "FPKM-TOM", verbose = 3) moduleLabelsAutomatic <- net$colors moduleColorsAutomatic <- labels2colors(moduleLabelsAutomatic) # A data frame with module eigengenes can be obtained as follows MEsAutomatic <- net$MEs
展示网络图:plotDendroAndColors(net$dendrograms[[1]], moduleColorsAutomatic[net$blockGenes[[1]]], "Module colors", dendroLabels = FALSE, hang = 0.03, addGuide = TRUE, guideHang = 0.05)
Example wgcna_module:- Mine:
2.5 关联模块与表型,作相关系数图
- 结合模块与样本性状的相关性来分析,将模块与表型关联:
nGenes <- ncol(dataExpr) nSamples <- nrow(dataExpr) #same to MEsAutomatic MEs0 <- moduleEigengenes(dataExpr, moduleLabelsAutomatic)$eigengenes #ME(module eigengene) MEs <- orderMEs(MEs0) modTraitCor <- cor(MEs, Traits, use = "p") modTraitP <- corPvalueStudent(modTraitCor, nSamples)
- 然后做模块与表型相关系数图:
textMatrix <- paste(signif(modTraitCor, 2), "\n(", signif(modTraitP, 1), ")",sep = "") dim(textMatrix) <- dim(modTraitCor) par(mar = c(6, 5.5, 3, 3)) labeledHeatmap( Matrix = modTraitCor, xLabels = names(Traits), yLabels = names(MEs), ySymbols = names(MEs), colorLabels = FALSE, colors = greenWhiteRed(50), textMatrix = textMatrix, setStdMargins = FALSE, cex.text = 0.7, zlim = c(-1, 1), main = paste("Module-trait relationships") )
- 图中,y轴是模块名,x轴是性状,每个模块都跟每个性状有一个空格,空格的上部分是模块与性状的相关系数,空格的下部分是一个p值,代表相关系数的显著性
其实随着 WGCNA 的广泛使用,其用处不仅局限于对性状的分析,还可以结合模块与样本的相关性来分析- 比如当研究不同处理组织或者不同发育时期的植物时,如果想了解处理或者不同时期中那些模块对其有显著影响,那么我们可以自行构建类似于性状的 Traits 矩阵
- 最简单的一种方法就是:默认为各个样本间是无关联的,那么Traits 矩阵则为标量矩阵,当然还有其他方式(比如考虑样品有生物学重复的情况)
2.6 根据模块间的相似性及向异性,作树杈图
- 查看每个模块之间的相似性以及向异性,并作树杈图
dissimME <- (1-t(cor(MEs, method="p")))/2 hclustdatME <- hclust(as.dist(dissimME), method="average" ) # Plot the eigengene dendrogram plot(hclustdatME, main="Clustering tree based of the module eigengenes")
- wgcna_hclust:
2.7 查看特定模块中的特定基因与特定形状关系
- 查看特定模块中的基因对于特定性状是否具有高GS值(gene significance)和MM值(module membership)
- 以 weigth 为例:
- 需要先计算每个基因在每个模块中的KME值
- 然后再计算GS值
- 最后作图
MEs0 <- moduleEigengenes(dataExpr, moduleColorsAutomatic)$eigengenes weight <- as.data.frame(Traits$weight_g) names(weight) = "weight" GS.weight = as.numeric(cor(dataExpr, weight, use = "p")) MEs <- orderMEs(MEs0) datKME <- signedKME(dataExpr, MEs) ME2Column <- substring(names(MEs), 0) column <- match("MEblack", ME2Column) restModule = moduleColorsAutomatic == "black" verboseScatterplot(MEs[restModule, column], GS.weight[restModule], xlab = paste("Module Membership ","black", "module"), ylab = "GS.weight", main = paste("kME.", "black", "vs. GS"), col = "black")
- wgcna_GC_MM:
2.8 最后导出模块中 TOM值 的数据,导入 cytoscape 作图
- 最后导出模块中TOM值的数据,导入 cytoscape 作图:
TOM <- TOMsimilarityFromExpr(dataExpr, power = 7) # Select modules需要修改,选择需要导出的模块颜色 modules <- c("black") # Select module probes probes <- colnames(dataExpr) inModule <- is.finite(match(moduleColorsAutomatic, modules)) modProbes <- probes[inModule] # Select the corresponding Topological Overlap modTOM <- TOM[inModule, inModule] dimnames(modTOM) <- list(modProbes, modProbes) # Export the network into edge and node list files Cytoscape can read cytoscape <- exportNetworkToCytoscape(modTOM, edgeFile = paste("FPKM-One-step-CytoscapeInput-edges-", paste(modules, collapse="-"), ".txt", sep=""), nodeFile = paste("FPKM-One-step-CytoscapeInput-nodes-", paste(modules, collapse="-"), ".txt", sep=""), weighted = TRUE, threshold = 0.2, nodeNames = modProbes, nodeAttr = moduleColors[inModule])
- 并做一个TOM图,如下:
dissTOM <- 1 - TOM plotTOM = dissTOM^7 diag(plotTOM) <- NA geneTree <- net$dendrograms[[1]] TOMplot(plotTOM, geneTree, moduleColorsAutomatic, main = "Network heatmap plot, all genes")
wgcna_TOM:
2.9 用 cytoscape 将上述特定模块中的基因以互作网络图形式展现,以一定的标准来选择 hub gene 作为后续研究的重点
- 也可以对模块中的每个基因进行注释,做GO以及KEGG富集
- 根据富集到的通路,结合模块信息来确定哪个模块的基因作为后续研究的重点


























 1203
1203











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











