









生物信息分析必备 | GSEA富集分析一文通透
1. 富集分析概述
基因富集分析(Gene Enrichment Analysis)是一种常用的生物信息学方法,用于解释在基因组或基因集合中出现的显著富集的功能或特定特征。这种分析用于高通量基因表达数据的解释,比如基因芯片数据或RNA测序数据。其通常包含以下四种功能注释分析:
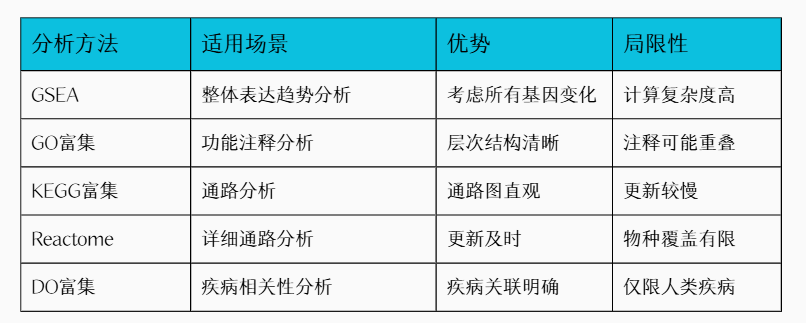
2. 富集分析数据库简介
不管想做哪种富集通路分析,均可去在线网站进行操作,下面列出了常用的富集通路数据库,本文主要介绍R代码实现通路富集分析,详细示例见目录章节 “5. 示例操作”
👉 温馨提示:R代码实操示例请直接下滑到文末 “5. 示例操作” 章节查看。
1. KEGG(Kyoto Encyclopedia of Genes and Genomes)
- 简介:是一个包含生物系统功能和信息的数据库资源,提供了关于基因组、化学物质、代谢途径、疾病和药物等方面的信息。KEGG数据库涵盖了多个生物学领域,包括基因组学、代谢组学、药物发现等,为研究人员提供了丰富的生物信息资源,用于理解生物系统的结构、功能和动态变化。
- 特点:
- 整合基因组、代谢通路、药物和疾病信息
- 覆盖几乎所有物种
- 提供详细的通路图
- 访问地址:https://www.kegg.jp/
2. GO(Gene Ontology)
- 简介:是一种用于描述基因功能和基因组学数据的标准化语言和分类体系。它是一个用于注释基因和蛋白质的功能、过程和细胞组件的分类系统。GO 分为三个主要的分类:分子功能(Molecular Function)、生物过程(Biological Process)和细胞组件(Cellular Component)。研究人员可以使用GO标准对基因和蛋白质进行分类和注释,从而更好地理解它们在生物学过程中的作用和相互关系。
- 三大分类:
- 生物学过程(Biological Process, BP)
- 分子功能(Molecular Function, MF)
- 细胞组件(Cellular Component, CC)
- 访问地址:http://geneontology.org/
3. Reactome(Reactome Pathway Database)
- 简介:Reactome 是一个开源、手工注释、经过同行评议的生物通路数据库。它提供了从小分子代谢到转录调控、信号转导和复杂的细胞过程等多个层面的生物学通路信息。
- 特点:
- 提供直观的通路可视化工具
- 包含30多个物种信息
- 定期更新维护
- 访问地址:https://reactome.org/
4. DO(Disease Ontology)数据库
- 简介:Disease Ontology 是一个标准化的疾病本体知识库,旨在提供人类疾病的统一命名和分类体系。它将人类疾病与基因组、变异信息、药物反应等多个生物医学领域关联起来。
- 特点:
- 标准化疾病术语
- 疾病表型特征描述
- 相关医学词汇整合
- 访问地址:http://www.disease-ontology.org/
3. GSEA的原理
GSEA的核心思想是将所有基因按照表达差异的显著性进行排序,然后检测特定基因集在这个排序中的富集程度。主要步骤包括:
- 基因排序:根据差异表达分析的统计量(如logFC或p值)对基因进行排序。
- 富集评分(Enrichment Score, ES):计算每个基因集在基因排序中的富集分数。
- 统计显著性评估:使用置换检验(Permutation Test)来评估富集分数的显著性。
- 多重检验校正:对显著性结果进行FDR校正,减少假阳性。
4. GSEA的分析步骤
- 数据准备:整理表达矩阵和分组信息。
- 基因集选择:选择合适的基因集(如KEGG、GO、Hallmark等)。
- 运行GSEA:使用GSEA工具对数据进行富集分析。
- 结果可视化:生成富集曲线、热图等图表。
- 生物学解释:对显著富集的通路或功能进行解读。
5. 示例操作
# 加载必要的包
library(ReactomePA)
library(tidyverse)
library(data.table)
library(org.Hs.eg.db) # 人类基因注释数据库
library(clusterProfiler) # GSEA核心包
library(enrichplot) # 富集分析可视化包
library(dplyr)
# 读取数据
gene_data <- read.csv("APT_vs_AHa.csv", header = TRUE, row.names = 1)
# 注意:输入文件必须包含log2FoldChange列
# 准备基因列表
gene_list <- data.frame(
Gene = rownames(gene_data),
Score = gene_data$log2FoldChange # 重要:这里使用log2FoldChange作为排序依据
) %>%
filter(is.finite(Score)) # 过滤掉无穷大值
# 转换基因ID
gene_ids <- mapIds(org.Hs.eg.db,
keys = gene_list$Gene,
column = "ENTREZID", # 转换为ENTREZID格式
keytype = "SYMBOL", # 输入为基因符号格式
multiVals = "first") # 对于多个匹配只取第一个
# 合并数据并创建排序的基因列表
gene_list$ENTREZID <- gene_ids
final_list <- gene_list %>%
filter(!is.na(ENTREZID)) %>%
arrange(desc(Score))
# 创建命名向量
gene_scores <- final_list$Score
names(gene_scores) <- final_list$ENTREZID
# 执行GSEA分析
# GO分析
go_result <- gseGO(gene_scores,
OrgDb = org.Hs.eg.db,
ont = "ALL", # 可选 "BP", "CC", "MF"
minGSSize = 10, # 最小基因集大小
maxGSSize = 500, # 最大基因集大小
pvalueCutoff = 0.05) # p值阈值
# KEGG分析
kegg_result <- gseKEGG(gene_scores,
organism = "hsa", # 人类的organism代码
minGSSize = 10,
maxGSSize = 500,
pvalueCutoff = 0.05)
# Reactome分析
reactome_result <- gsePathway(gene_scores,
organism = "human",
minGSSize = 10,
maxGSSize = 500,
pvalueCutoff = 0.05)
# 保存结果
write.csv(as.data.frame(go_result), "test_analysis_GO.csv", row.names = FALSE)
write.csv(as.data.frame(kegg_result), "test_analysis_KEGG.csv", row.names = FALSE)
write.csv(as.data.frame(reactome_result), "test_analysis_Reactome.csv", row.names = FALSE)
# 绘图
pdf("GSEA_plots.pdf", width = 10, height = 8) # 可根据需要调整图片大小
# GO富集分析图
dotplot(go_result, showCategory = 20) + ggtitle("GO Enrichment Analysis")
# KEGG通路富集分析图
ridgeplot(kegg_result, showCategory = 15) + ggtitle("KEGG Pathway Enrichment")
# Reactome通路富集分析图
dotplot(reactome_result, showCategory = 20) + ggtitle("Reactome Pathway Enrichment")
dev.off()
6. GSEA结果解读
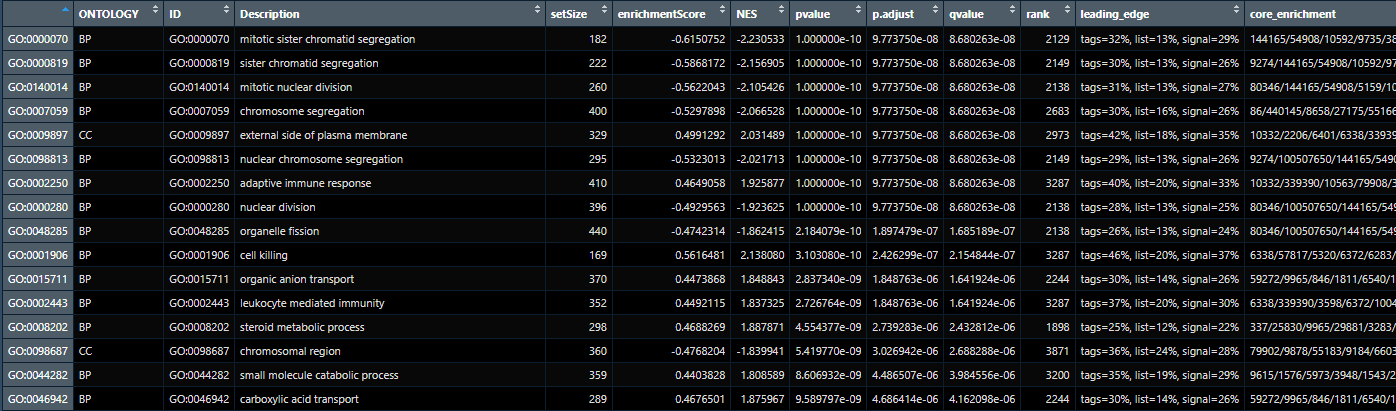
在分析GSEA结果时,我们最需要关注的几列是:
p.adjust(校正后的P值)、NES(标准化富集分数)和Description(功能描述)。这三列能直观地告诉我们哪些通路显著富集,以及富集的方向和强度。
完整的结果文件中,每一列的具体含义如下:
- Description & ID: GO功能描述及其唯一标识号
- ONTOLOGY: GO的三大类别(BP生物学过程、CC细胞组分、MF分子功能)
- enrichmentScore: 富集分数
- NES: 标准化富集分数,正值表示正向富集,负值表示负向富集
- pvalue: 原始P值
- p.adjust: 多重检验校正后的P值,通常<0.05认为显著富集
- qvalue: FDR校正值
- setSize: 该通路包含的基因数量
- leading_edge: 前沿分析结果(tags/list/signal的百分比)
- core_enrichment: 核心富集基因列表
小贴士:解读时建议先看 p.adjust 和 NES ,找出显著富集的通路,再结合Description了解具体的生物学意义哦!😊
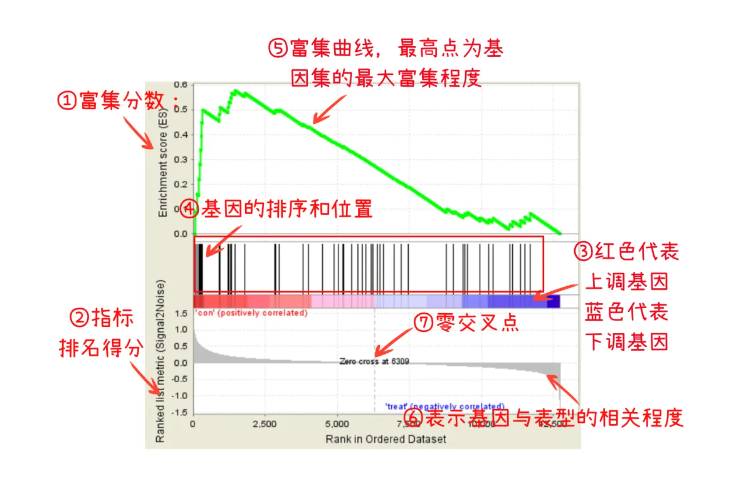
① 富集分数(Enrichment Score):表示基因集富集程度的量化指标。
② 指标排名得分(Ranking Metric Score):用于基因排序的统计量。
③ 基因表达变化区域:红色区域表示基因表达上调,蓝色区域表示基因表达下调,反映了基因在不同条件下的表达变化趋势。
④ 基因位置标记(Hit):黑色线条展示基因集中每个基因出现在基因排序列表中的位置,每一根线条代表基因集中的一个分子,可以看出哪些基因在富集评分中贡献最大。
⑤ 富集曲线(Enrichment Plot):绿色曲线表示ES评分的累积过程,其峰值表示基因集的最大富集程度。峰值为正表示富集通路上调,峰值为负表示富集通路下调。
⑥ 基因排名指标得分:表示基因与表型的相关程度,零交叉点是从正变为负的位置,帮助区分正/负相关的基因。
7. GSEA在生物医学研究中的应用
- 疾病机制解析:揭示疾病相关的通路和分子机制。
- 药物作用机制研究:探索药物治疗的潜在通路。
- 生物标志物发现:识别与特定疾病状态相关的基因集。
8. 总结
GSEA作为一种强大的基因集富集分析工具,在揭示复杂生物学过程和疾病机制中发挥着重要作用。通过合理选择基因集和工具,可以深入理解基因表达数据背后的生物学含义,为科研工作提供有力支持。
9. 联系方式
好啦,今日分享毕!更多科研干货、绘图技能关注 N今天C了么 不迷路!



















 4583
4583

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









