






体细胞短变体检测 (SNV + InDel)
Somatic short variant discovery (SNVs + Indels)
目的
在单个个体的一个或多个肿瘤样本中,识别体细胞短变异(SNV和InDel),无论是否有匹配的正常样本(With or without a matched normal sample)。
参考实现
管道 | 总结 | 笔记 | Github | Terra |
体细胞短变异体Tumor-Normal配对 | T-N Bam到VCF | 通用 | 有 | b37 |
体细胞短变异PON的生成 | 正常样本Bam到PON | 通用 | 有 | b37 |
另见文件:
gatk-master.zip
(How to) Call somatic mutations using GATK4 Mutect2.docx
链接:https://pan.baidu.com/s/1BTmmCM-mJ-hEA1wWNYDs4g
提取码:ysx4
预期的输入
该工作流程需要为每个输入的肿瘤和正常样本(Each input tumor and normal sample)提供BAM文件。输入Bam应按照《GATK数据预处理最佳实践 (GATK Best Practices for data pre-processing)》中的描述进行预处理:

https://gatk.broadinstitute.org/hc/en-us/articles/360035535912
体细胞短变异检测最佳流程实践图 (Mutect2)
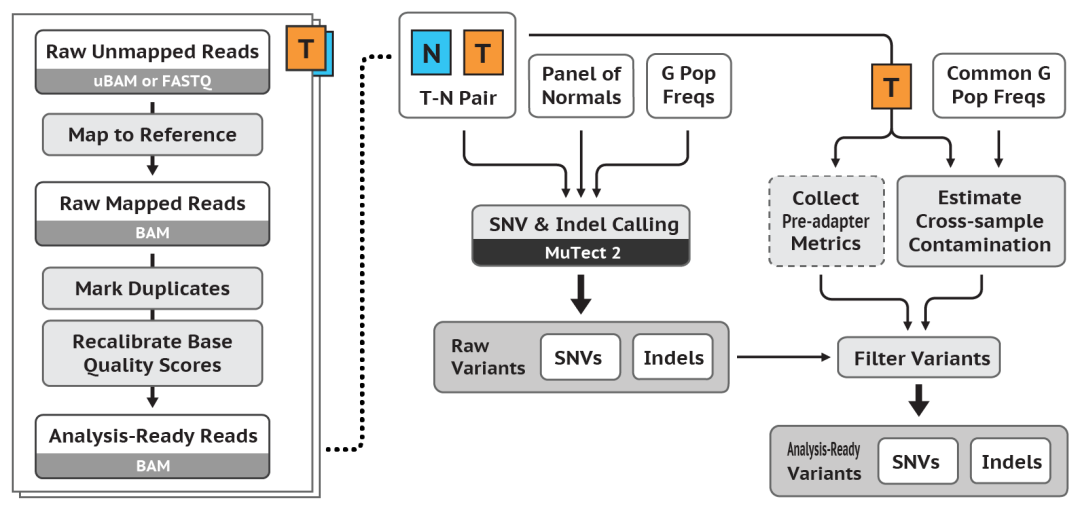
主要步骤
这个工作流有两个主要步骤:① 首先,生成大量的候选体细胞变异;② 然后,对它们进行筛选,以获得更有信心的体细胞变异调用集合 (A more confident set of somatic variant calls)。
检测候选变异(Call candidate variants)
工具:Mutect2
https://gatk.broadinstitute.org/hc/en-us/articles/5358824293659--Tool-Documentation-Index#Mutect2
与HaplotypeCaller一样,Mutect2通过激活区内的单倍型的局部de-novo组装 (Local de-novo assembly of haplotypes in an active region),同时调用(Call)SNV和InDel,最终由Bam文件(+参考基因组)获得VCF文件。
也就是说,当Mutect2遇到一个显示出体细胞变异迹象的区域时,它会丢弃现有的映射/比对信息,并完全重新组装该区域的Reads (Completely reassembles the reads in that region),以生成候选的变异单倍型 (Candidate variant haplotypes)。
与HaplotypeCaller一样,Mutect2随后会通过Pair-HMM算法将每个Read与每个单倍型进行比对 (Aligns each read to each haplotype),从而获得一个可能性矩阵。最后,它应用贝叶斯体细胞可能性模型 (Bayesian somatic likelihoods model)来获得等位基因为体细胞变异相对于测序错误的对数几率 (Obtain the log odds for alleles to be somatic variants versus sequencing errors)。
非遗传性肿瘤测序数据分析的难点
不像胚系突变(人体中所有细胞的遗传物质、基因型、突变型基本一致),体细胞突变的取样和测序数据生信分析都有一定的难度。① 取样:需要尽量获取受累组织(需要仔细甄别细胞外观,甚至生化检测结果),否则如果肿瘤细胞占比很极低时,将影响整个数据的分析结果,甚至导致错误的结论;② 数据分析:在取样没有问题时才可进行(极大地受到肿瘤细胞占比等因素影响)。即使肿瘤占比较高,但是由于肿瘤突变的异质性很强,许多与肿瘤相关的突变的突变频率极低,甚至低于测序仪的检测错误率。因此,肿瘤WES分析通常要求能够准确检测到低至0.5% ~ 10%的突变丰度,这就是体细胞分析最大的难点,也是与胚系突变检测的最大不同(胚系突变一旦发生,对于人类等二倍体生物来说,就至少是杂合子,即至少50%的理论突变率)。
因此,整个非遗传性肿瘤(即由于后天的基因突变引起的肿瘤)的体细胞检测流程一直在克服以下几个问题:① 样本交叉污染;② PCR扩增等建库过程中引入的“假阳性”突变;③ 测序仪引入的测序错误 (约1%,测序仪的真实测序碱基质量值非常重要);④ 病人自身携带的先天遗传突变 (生下来就有的、与参考基因组所不同的碱基序列),需过滤自身正常组织或癌旁组织的突变;⑤ 需过滤克隆性造血过程中自身白细胞等细胞中携带的突变;⑥ 过滤掉其它与肿瘤无关的体细胞突变。最终才是与肿瘤有关的体细胞突变。
计算污染
涉及的工具:GetPileupSummaries, CalculateContamination
https://gatk.broadinstitute.org/hc/en-us/articles/5358824293659--Tool-Documentation-Index#GetPileupSummaries
https://gatk.broadinstitute.org/hc/en-us/articles/5358824293659--Tool-Documentation-Index#CalculateContamination
该步骤对每个肿瘤样本中存在交叉样本污染 (Cross-sample contamination)的Reads的分数/比例进行估计,并对每个肿瘤样本的等位基因拷贝数分段(Allelic copy number segmentation)进行估计。
与其它污染计算工具不同,CalculateContamination被设计成即使在有显著拷贝数变异的样本中,也可以在没有匹配正常样本 (Without a matched normal)的情况下很好地工作,并且对污染样本的数量不做任何假设。
Learn Orientation Bias Artifacts
工具:LearnReadOrientationModel
该工具使用Mutect2的可选F1R2计数输出(F1R2 counts output)来学习定向偏差模型(A model for orientation bias)的参数。它在为每个三核苷酸上下文 (Each trinucleotide context)排序之前,找到单链替换错误的先验概率 (Prior probabilities)。这对于FFPE肿瘤样本是极其重要的。
过滤变异 (Filter Variants)
工具包括:FilterMutectCalls
https://gatk.broadinstitute.org/hc/en-us/articles/5358824293659--Tool-Documentation-Index#FilterMutectCalls
Mutect2的体细胞可能性模型(Somatic likelihoods model)假设Read的错误是独立发生的,因此,例如,4个Reads,每1个Read的错误概率为1/1000,产生大约1000^4的对数几率 (A log odds of roughly 1000^4),有利于其成为真正的变异,而不是测序错误。
FilterMutectCalls解释了相关的错误,也就是说,一个位点上所有的变异Reads都是由于某种共同的错误来源 (Due to some common source of error)的可能性。它通过几个硬过滤 (Several hard filters)来实现这一目标,以检测:“Alignment artifacts and probabilistic models for strand and orientation bias artifacts”(比如PCR产生的错误,会被后续的PCR循环传递下去,就出现“某种共同的错误来源”)、聚合酶滑移(Slippage artifacts,)、胚系变异和污染(都会出现“某种共同的错误来源”)。原因是:肿瘤体细胞突变相对随机,且通过超声法破碎后Reads长度大小不一。也因为如此,克隆性造血过程中自身白细胞等细胞中携带的突变,以及其它与肿瘤无关的体细胞突变将无法被过滤(除非做了患者自身配对的WBC实验设计及测序),因为这类突变也是:“相对随机,且通过超声法破碎后Reads长度大小不一”。
此外,FilterMutectCalls学习了一个关于肿瘤的整体SNV和InDel突变率和等位基因分数谱的贝叶斯模型,以改进Mutect2发出的对数几率 (A Bayesian model for the overall SNV and indel mutation rate and allele fraction spectrum of the tumor to refine the log odds emitted by Mutect2)。然后,它会自动设置一个过滤阈值,以优化:F评分、灵敏度和精度的调和平均值(F score, the harmonic mean of sensitivity and precision)。
注释变体
工具包括:Funcotator
https://gatk.broadinstitute.org/hc/en-us/articles/5358824293659--Tool-Documentation-Index#Funcotator
在这一步,我们运行一些工具,向数据集中发现的变异添加信息,比如:该突变发生在哪个基因,哪个外显子,氨基酸如何变化等。
其中一个工具是Funcotator,可以用来给每个变异添加基因级 (Gene-level)信息。
Funcotator是核心GATK工具集(Core GATK toolset)中的一个功能性注释工具,被设计用来处理体细胞和胚系的使用案例。
Funcotator在VCF文件中读取数据,用23种不同的变异分类(Variant classifications)中的1种来标记每个变异,产生基因信息(例如受影响的基因、预测的变异氨基酸序列等),以及与数据源中的信息的关联。
Funcotator支持的数据源包括GENCODE(基因信息和蛋白质变化预测)、dbSNP、gnomAD和COSMIC(等等)。数据源的语料库(The corpus of datasources)是可扩展的,用户可配置的,包括谷歌云存储支持的基于云的数据源。
Funcotator生成一个“变异调用格式(Variant Call Format, VCF)”文件(在INFO字段中增加注释结果)或一个“变异注释格式(Mutation Annotation Format, MAF)”文件。
一些问题反馈
上面文字里的GetPileupSummaries和CalculateContamination怎么用还不清楚。应该先使用哪个工具?或者它们应该同时使用吗?
这个管道能否用于在单细胞ATAC-seq或单细胞RNA-seq数据上调用SNV + InDel?
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|




























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集



















 548
548

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









