










我们接上文:满分室间质评之GATK Somatic SNV+Indel+CNV+SV一文中实现了对于卫计委室间质评数据分析以及与满分结果的匹配。本文将着重解决,保证最终结果一致的情况下,如何优化分析性能(并行化),如何将分析时间从 3h 59m 53s缩短至 1h 10m 38s。
- 优化的方向:实际运行GATK4.X的工具如Mutect2时,发现其运行效率相当低,从CPU占用率,内存占用,硬盘I/O都占用很低,起初自己DIY时候,将要分析的bed/interval_list文件按照染色体编号拆分(不太确定分析结果的一致性,所以比较谨慎),然后并行分析,最后将结果合并。后来GATK从4.0.6.0升级到4.1.3.0时候发现官方的best practice pipeline也做了类似的处理,这里就有了优化的空间。
- 还有一些工具Cnvkit,Manta,samtools depth 和 samtools flagstat 运行时对硬件资源利用也不充分(CPU占用,内存、硬盘IO等),可以考虑把这些任务并行运行以减少最终的运行时间。
- 优化后结果的一致性,首先官方提供了一系列工具,从直接感觉上应该是没有问题的,从室间质评的结果来看,标准结果上的突变一致性没有问题。非标准结果上会有一些出入,不影响最终结果。当然,目前还做不到全自动,最终的结果还是要使用IGV人工检查一遍。
本文中分析流程(pipeline)的运行环境
| 名称 | 描述 | 数量 |
|---|---|---|
| CPU | AMD Ryzen3 3950X 16核心32线程 全核满载4.2G | ×1 |
| 内存 | 64G | 16G ×4 |
| 硬盘 | 海康威视 C2000Pro 2T | ×1 |
| 三星PM983 3.84T | ×1 |
本文用到的分析流程文件及结果
| 名称 (点击下载) | 备注 |
|---|---|
| FFPE SNV CNV SV V2.4.workflow | 分析流程文件,可以一键导入SliverWorkspace分析系统(点击查看操作) 当然可以参照图片中运行脚本,shell里运行,效果也是一样 |
| 最终结果过滤脚本(python2.7 )及编译版本 | Illumina_pt2.bed 等用到的bed,intelval等文件 SnvAnnotationFilter.py SNV过滤脚本 CnvAnnotationFilter.py CNV过滤脚本 SvAnnotationFilter.py SV 过滤脚本 QcProcessor.py 获取整体QC数据的脚本 report_template.docx 分析报告模板 |
| 分析结果(pipeline结果与标准答案) | result.zip pipeline结果与标准答 |
本文用到的环境变量
| 变量名称 | 变量值 | 类型 |
|---|---|---|
| sn | 1701 | 数值 |
| data | /opt/data (原始fastq文件存放目录) | 目录 |
| result | /opt/result(中间文件及最终结果文件存放目录) | 目录 |
| tools.java | /opt/jdk1.8.0_162/bin/java | 程序 |
| tools.fastqc | /opt/FastQC/fastqc | 程序 |
| tools.bwa | /opt/bwa/bwa | 程序 |
| tools.samtools | /opt/samtools/samtools | 程序 |
| tools.sambamba | /opt/ref/sambamba-0.7.0-linux-static | 程序 |
| tools.bgzip | /opt/tabix-0.2.6/bgzip | 程序 |
| tools.tabix | /opt/tabix-0.2.6/tabix | 程序 |
| tools.gatk | /opt/ref/gatk-4.1.3.0/gatk | 程序 |
| tools.manta | /opt/manta/dist/bin/configManta.py | 程序 |
| tools.cnvkit | /usr/local/bin/cnvkit.py | 程序 |
| refs.gene | /opt/ref/hg19_refGene.txt | 文件 |
| refs.dict | /opt/ref/ucsc.hg19.dict | 文件 |
| refs.hum | /opt/ref/ucsc.hg19.fa | 字符 |
| refs.bed | /opt/ref/projects/Illumina_pt2.bed | 文件 |
| refs.interval | /opt/ref/projects/Illumina_pt2.interval_list | 文件 |
| refs.dbsnp | /opt/ref/dbsnp_138.hg19.vcf | 文件 |
| refs.mills | /opt/ref/Mills_and_1000G_gold_standard.indels.hg19.vcf | 文件 |
| refs.1000G | /opt/ref/1000G_phase1.indels.hg19.vcf | 文件 |
| refs.af_only | /opt/ref/af-only-gnomad.raw.sites.hg19.vcf.gz | 文件 |
| refs.small.exac | /opt/ref/small_exac_common_3_b37.vcf | 文件 |
| envis.threads | 32 | 数值 |
| cutoff.TLOD | 16.00 | 数值 |
| cutoff.event | 2 | 数值 |
| cutoff.cnvdep | 1000 | 数值 |
| cutoff.nvaf | 0.005 | 数值 |
| cutoff.cnv_min | -0.5 | 数值 |
| cutoff.cnv_max | 0.5 | 数值 |
| cutoff.vaf | 0.01 | 数值 |
| envis.scatter | 8 | 数值 |
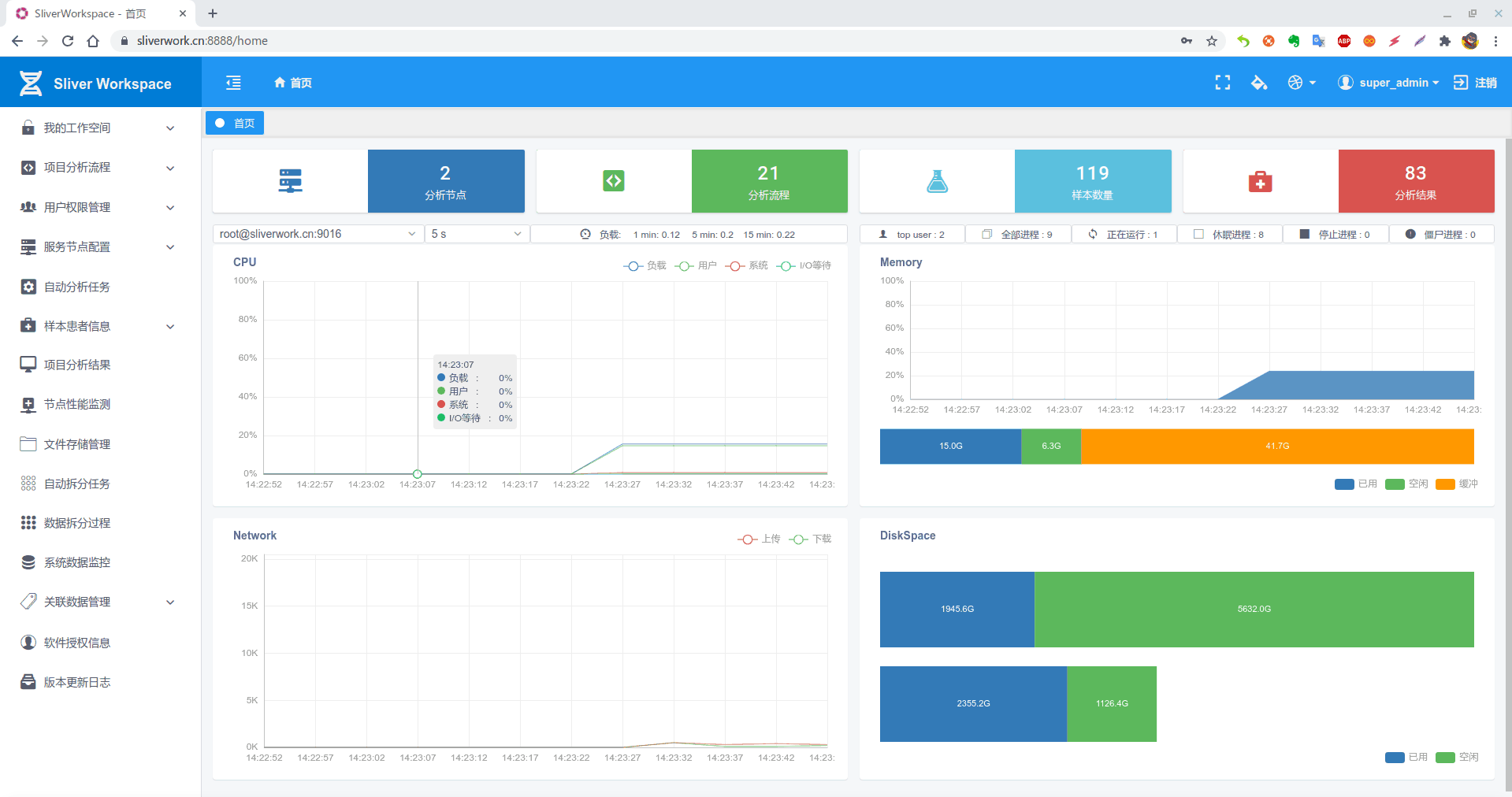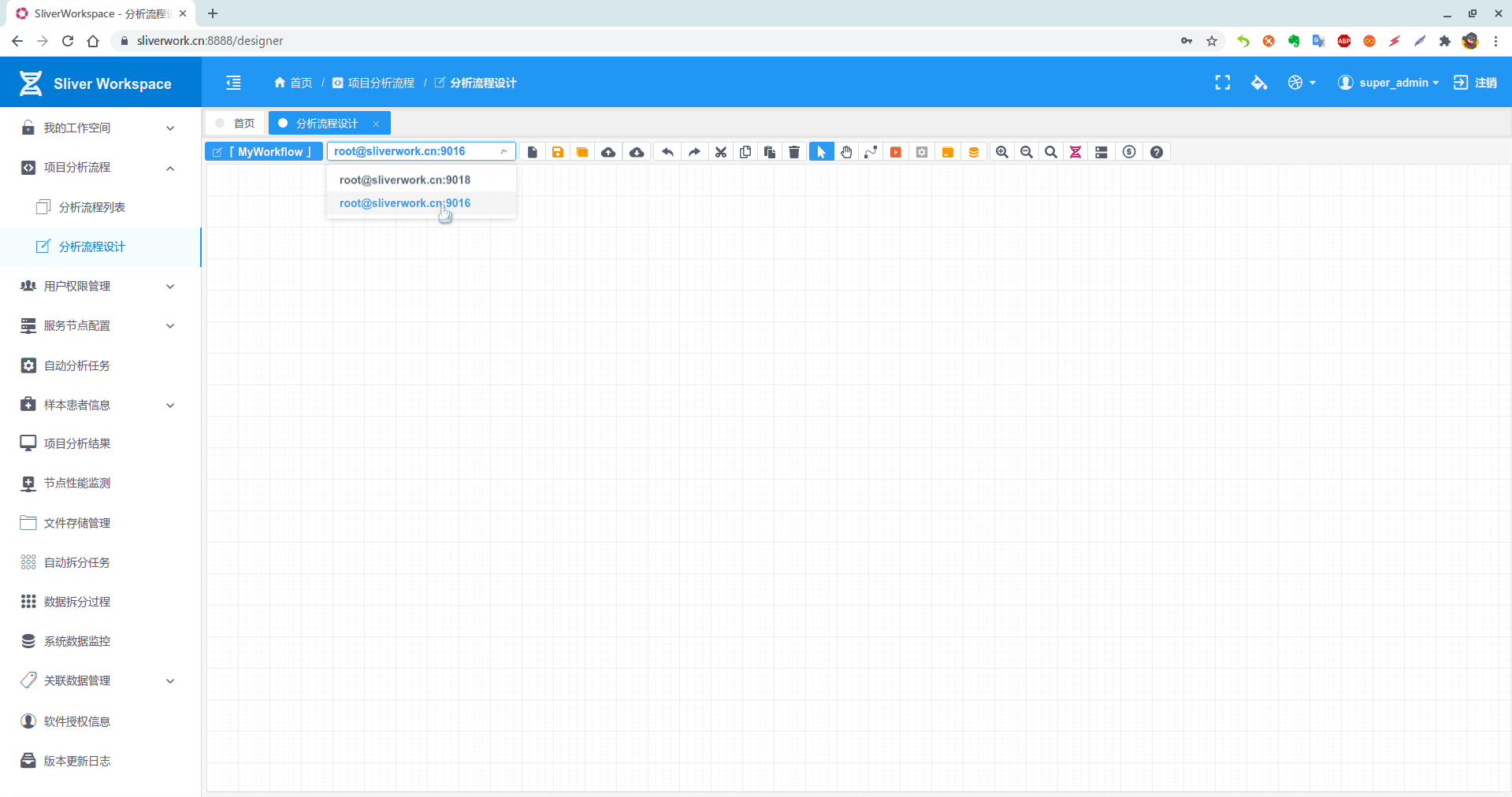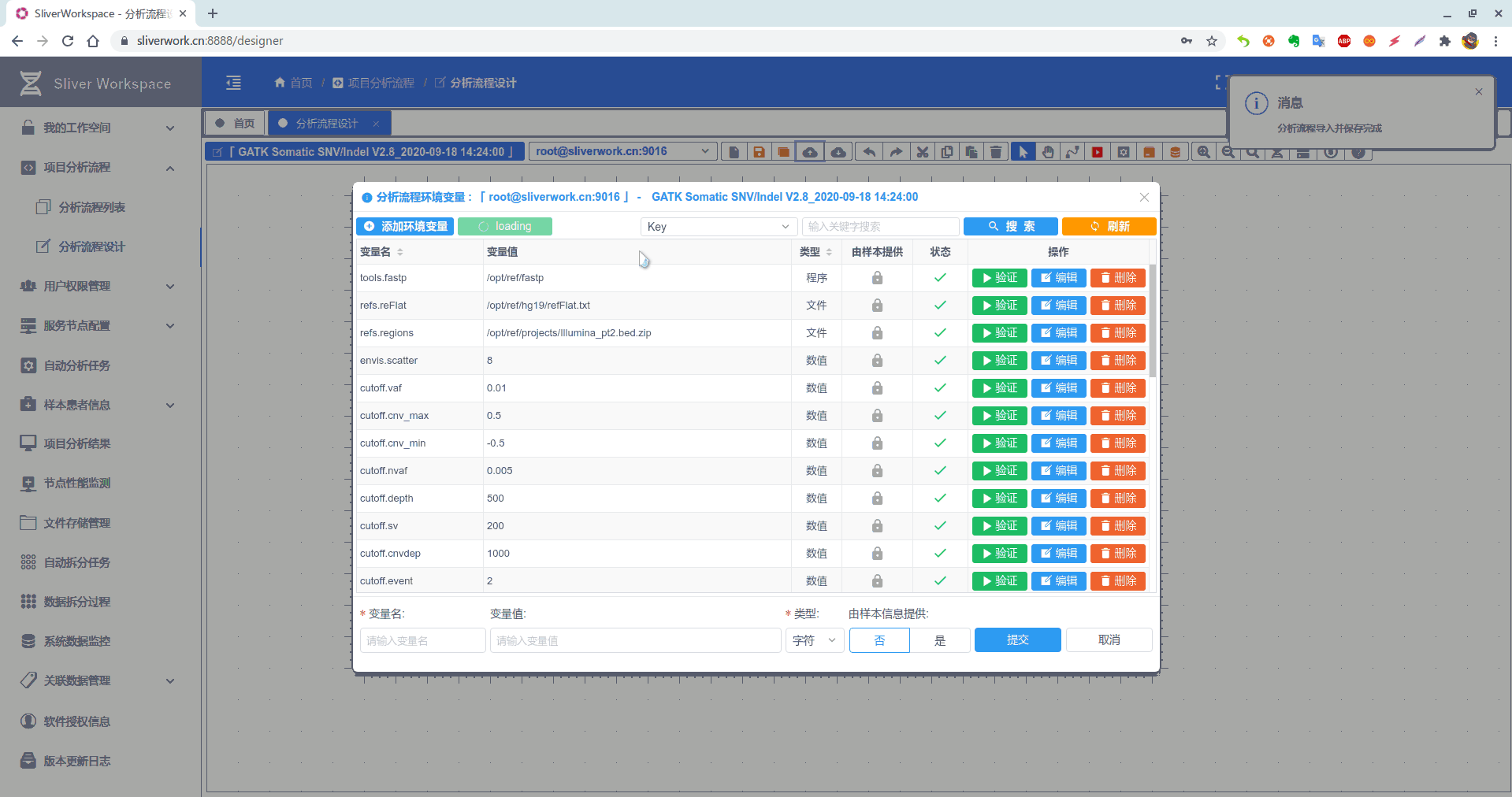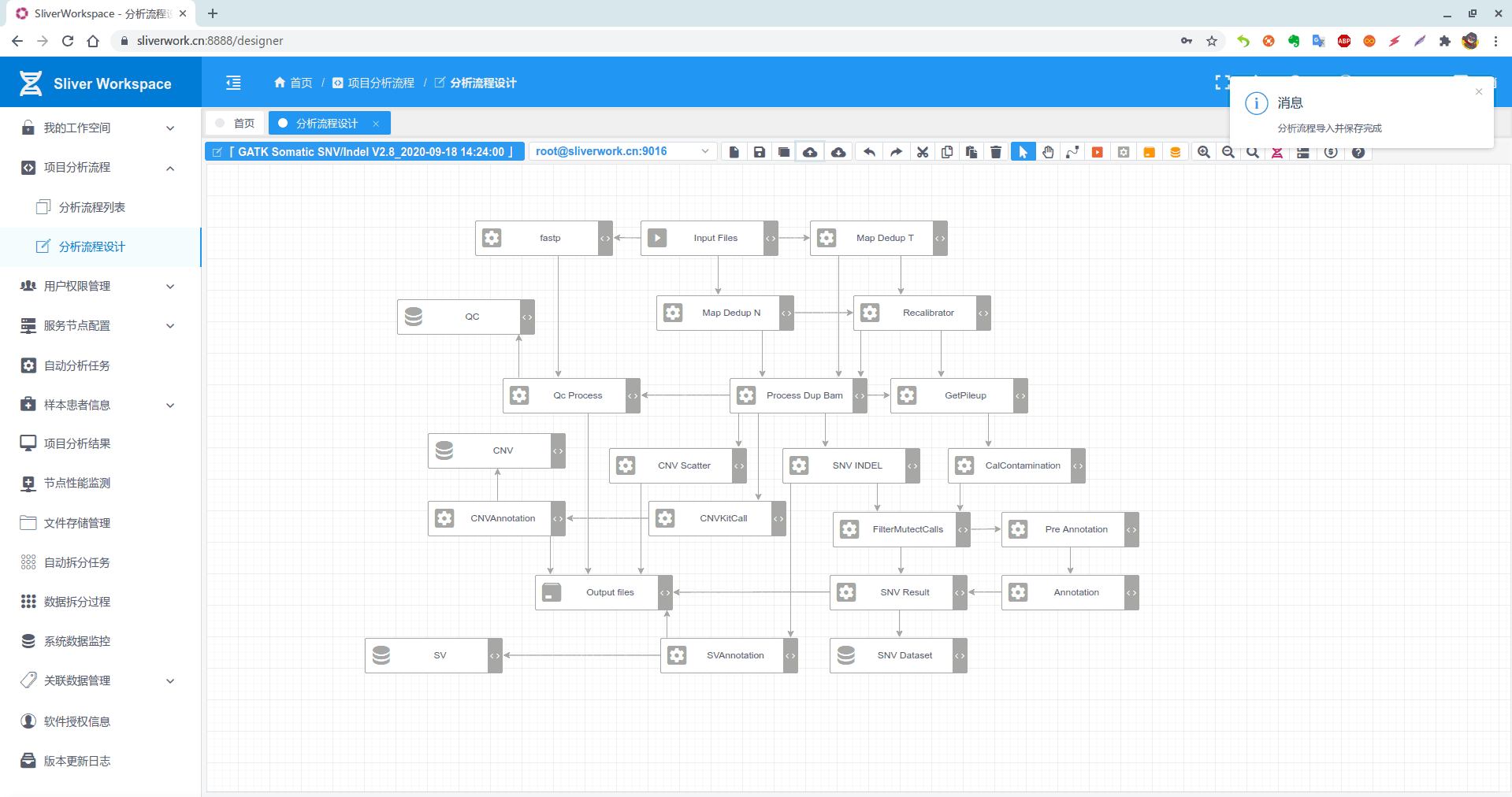
惯例:优化后分析流程(pipeline):

下面看看优化过程
- INPUT 输入文件(无变化)

- Normal:Map & Order & MarkDuplicate 合并完成

#bwa map完成接管道操作sambamba转换为bam,然后管道操作sambamba排序
${tools.bwa} mem \
-t ${envis.threads} -M \
-R "@RG\\tID:${sn}NC\\tLB:${sn}NC\\tPL:Illumina\\tPU:Miseq\\tSM:${sn}NC" \
${refs.hum} ${data}/${sn}NC_R1.fastq.gz ${data}/${sn}NC_R2.fastq.gz \
| ${tools.sambamba} view -S -f bam -l 0 /dev/stdin \
| ${tools.sambamba} sort -t ${envis.threads} -m 2G --tmpdir=${result} -o {result}/${sn}NC_sorted.bam /dev/stdin
#设置每个进程打开文件的最大数为10240,防止markdup时候sambamba报错退出
ulimit -n 10240
#使用sambamba对sorted.bam标记重复
${tools.sambamba} markdup \
--tmpdir ${result} \
-t ${envis.threads} ${result}/${sn}NC_sorted.bam ${result}/${sn}NC_marked.bam
#sambamba生成的索引文件名与GATK默认的索引文件名不一致,这里重命名一下以符合GATK习惯
mv ${result}/${sn}NC_marked.bam.bai ${result}/${sn}NC_marked.bai
#删除${sn}NC_sorted.bam删除中间文件,节省硬盘空间
rm -f ${result}/${sn}NC_sorted.bam
- Tumor:Map & Order & MarkDuplicate 合并完成

除了fastq文件不同,处理过程一模一样
-
Recalibrator:Normal&Tumor 并行完成

-
Process Dup Bam (对于标记重复后的Bam文件的处理,合并了很多步骤)

```shell
#此处是原先Manta分析SV的步骤一,生成runWorkflow.py,因为这一不步速度很快,所以串行执行
rm -f ${result}/${sn}/runWorkflow.py
python ${tools.manta} \
--normalBam ${result}/${sn}NC_marked.bam \
--tumorBam ${result}/${sn}_marked.bam \
--referenceFasta ${refs.hum} \
--exome \
--callRegions /opt/ref/projects/Illumina_pt2.bed.zip \
--runDir ${result}/${sn}
# 对bam文件碱基质量校正的第二步,Normal & Tumor并行处理
${tools.gatk} ApplyBQSR \
--bqsr-recal-file ${result}/${sn}_recal.table \
-L ${refs.interval} \
-R ${refs.hum} \
-I ${result}/${sn}_marked.bam \
-O ${result}/${sn}_bqsr.bam &
${tools.gatk} ApplyBQSR \
--bqsr-recal-file ${result}/${sn}NC_recal.table \
-L ${refs.interval} \
-R ${refs.hum} \
-I ${result}/${sn}NC_marked.bam \
-O ${result}/${sn}NC_bqsr.bam &
#原先QC步骤,获取insert size,Normal & Tumor并行
${tools.gatk} CollectInsertSizeMetrics \
-I ${result}/${sn}_marked.bam \
-O ${result}/${sn}_insertsize_metrics.txt \
-H ${result}/${sn}_insertsize_histogram.pdf &
${tools.gatk} CollectInsertSizeMetrics \
-I ${result}/${sn}NC_marked.bam \
-O ${result}/${sn}NC_insertsize_metrics.txt \
-H ${result}/${sn}NC_insertsize_histogram.pdf &
# 运行manta SV分析
python ${result}/${sn}/runWorkflow.py -m local -j ${envis.threads} &
# 运行cnvkit CNV分析
${tools.cnvkit} batch \
${result}/${sn}_marked.bam \
--normal ${result}/${sn}NC_marked.bam \
--method hybrid \
--targets ${refs.bed} \
--annotate /opt/ref/refFlat.txt \
--output-reference ${result}/${sn}_reference.cnn \
--output-dir ${result}/ \
--diagram \
-p 0 &
#samtools统计测序深度
${tools.samtools} depth -b ${refs.bed} ${result}/${sn}_marked.bam > ${result}/${sn}_marked.depth &
${tools.samtools} depth -b ${refs.bed} ${result}/${sn}NC_marked.bam > ${result}/${sn}NC_marked.depth &
#samtools统计比对信息
${tools.samtools} flagstat --threads ${envis.threads} ${result}/${sn}_marked.bam > ${result}/${sn}_marked.flagstat &
${tools.samtools} flagstat --threads ${envis.threads} ${result}/${sn}NC_marked.bam > ${result}/${sn}NC_marked.flagstat &
#使用GATK Splitintervals工具将interval_list拆分成若干份。方便后面使用
#这里要讲讲从GATK4.1.3.0这个版本开始的骚操作了。我算法资源使用效率低是吧,我把interval文件拆分成几份,并行分析之后再把结果合并,来达到提高效率的目的。后面GetPileupSummaries和Mutect2都会用到。
#根据硬件性能决定拆分多少份,也就是并行多少个,我这里是8份
rm -f ${result}/${sn}/*.interval_list
${tools.gatk} SplitIntervals \
-R ${refs.hum} \
-L ${refs.interval} \
-O ${result}/${sn} \
--scatter-count ${envis.scatter}
wait
```
- Qc Process 处理以上文件,获取QC信息

- GetPileupSummaries

这里要讲讲GATK4.1.3.0这个版本开始的骚操作了。我算法资源使用效率低是吧,我把interval文件拆分成几份,并行分析之后再把结果合并,来达到提高效率的目的。
# 这里循环拆分的interval_list文件运行GetPileupSummaries
for i in `ls ${result}/${sn}/*.interval_list`;
do
${tools.gatk} GetPileupSummaries \
-R ${refs.hum} \
-I ${result}/${sn}_bqsr.bam \
-O ${i%.*}-pileups.table \
-V ${refs.small.exac} \
-L $i \
--interval-set-rule INTERSECTION &
${tools.gatk} GetPileupSummaries \
-R ${refs.hum} \
-I ${result}/${sn}NC_bqsr.bam \
-O ${i%.*}-pileups.nctable \
-V ${refs.small.exac} \
-L $i \
--interval-set-rule INTERSECTION &
done
wait
#将运行结果*.table文件作为参数合并成一行,运行GatherPileupSummaries将结果合并成一个
tables=
for i in `ls ${result}/${sn}/*.table`;
do
tables="$tables -I $i"
done
${tools.gatk} GatherPileupSummaries \
--sequence-dictionary ${refs.dict} \
$tables \
-O ${result}/${sn}_pileups.table
nctables=
for i in `ls ${result}/${sn}/*.nctable`;
do
nctables="$nctables -I $i"
done
${tools.gatk} GatherPileupSummaries \
--sequence-dictionary ${refs.dict} \
$nctables \
-O ${result}/${sn}NC_pileups.table
- CalculateContamination 计算污染

- Call SNV INDEL

#vcf-file.list记录了并行分析输出的结果,后面合并要用到
rm -f ${result}/${sn}/vcf-file.list
touch ${result}/${sn}/vcf-file.list
#循环使用拆分后的interval_list文件运行Mutect2
for i in `ls ${result}/${sn}/*.interval_list`;
do
rm -f ${i%.*}_bqsr.vcf.gz
${tools.gatk} Mutect2 \
-R ${refs.hum} \
-I ${result}/${sn}_bqsr.bam -tumor ${sn} \
-I ${result}/${sn}NC_bqsr.bam -normal ${sn}NC \
-L $i \
-O ${i%.*}_bqsr.vcf.gz \
--germline-resource ${refs.af_only} \
--native-pair-hmm-threads ${envis.threads} &
echo ${i%.*}_bqsr.vcf.gz >> ${result}/${sn}/vcf-file.list
done
wait
#生成合并参数,运行MergeMutectStats将状态文件合并
rm -f ${result}/${sn}_bqsr.vcf.gz.stats
stats=
for z in `ls ${result}/${sn}/*_bqsr.vcf.gz.stats`;
do
stats="$stats -stats $z"
done
${tools.gatk} MergeMutectStats $stats -O ${result}/${sn}_bqsr.vcf.gz.stats
#合并并行分析得到的vcf.gz
${tools.gatk} MergeVcfs \
-I ${result}/${sn}/vcf-file.list \
-O ${result}/${sn}_bqsr.vcf.gz
- FilterMutectCalls 使用GATK提供的过滤器过滤SNV&Indel

- 将过滤后的文件转换为Annovar注释所需要的格式

- 使用Annovar注释

- 使用自己写的脚本对注释后的结果过滤,比如按照室间质评要求,过滤掉突变频率低于1%,测序深度低于500的突变。对GATK某些过滤器过滤掉的结果进行保留和排除,后面使用IGV进行人工筛选。最终输出的结果为,${sn}.result.SNV.xls(其实是个csv文件,扩展名改为.xls是便于使用excel打开,很多人都这么干)

- 使用CnvKIt,获取CNV突变(接Process Dup Bam)

- 使用py脚本文件,对CnvKit输出结果过滤。同样根据hg19_refGene.txt文件匹配基因,以及发生拷贝数变异的区域的外显子区域等。

- 使用CnvKit画图

- 使用python脚本对Manta获取的SV过滤。如根据SOMATICSCORE分数过滤,根据hg19_refGene.txt提供文件,计算突变基因等等。(接Process Dup Bam)

- 最终结果

- 整个pipeline运行情况,可以看到消耗时间明显降低,约为原先1/3时间。
















 698
698

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言










{kind=link}