






点击蓝字 关注我们
多组学方法在肿瘤微生物组研究中的应用

原文链接DOI: https://doi.org/10.1002/imt2.73
综 述
● 2023年1月9日,俄亥俄州立大学郑庆飞团队在iMeta在线发表了题为“Applying multi-omics towards tumor microbiome research”的文章。
● 多组学(尤其是单细胞组学)将对未来微生物与肿瘤微环境相互作用的研究产生巨大影响。本文系统总结了多组学的研究进展及其在肿瘤微生物组研究中的现有和潜在应用,为研究者提供了一个可供参考的组学工具箱。
● 第一作者:张楠
● 通讯作者:郑庆飞(Qingfei.Zheng@osumc.edu)
● 合作作者:Shruthi Kandalai、周小状、Farzana Hossain
● 主要单位:俄亥俄州立大学医学院放射肿瘤学系、俄亥俄州立大学詹姆斯综合癌症中心癌症代谢中心、俄亥俄州立大学医学院生物化学和药理学系
亮 点
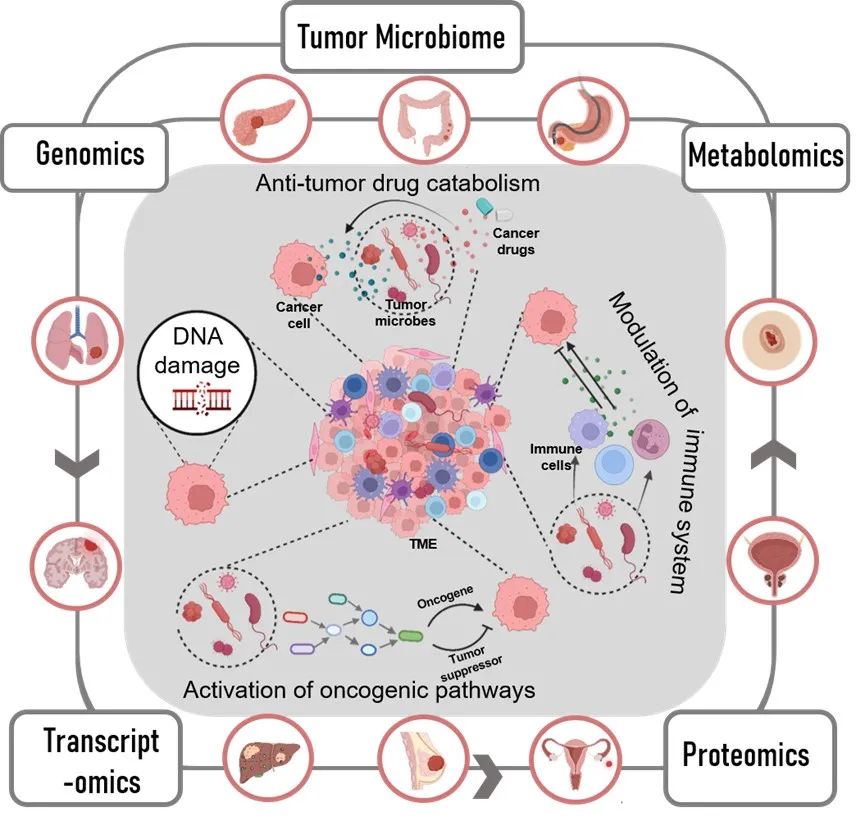
● 肿瘤微生物组在癌症发生、发展、转移和治疗中扮演着重要角色
● 肿瘤微生物组具有成为癌症早期诊断和治疗靶标的巨大潜力
● 多组学方法(基因组学、转录组学、蛋白质组学和代谢组学)是研究肿瘤微生物组的有力手段,促进了在中心法则不同水平的理解
● 多组学将对肿瘤微生物组的未来研究产生巨大影响
● 单细胞多组学技术的发展将推动肿瘤微生物组领域的进步
摘 要
肿瘤微生物群不是人体的“短期租户”,而是作为“永久居民”在癌症的发生、发展、转移和治疗中扮演着重要角色。由于肿瘤微生物组具有成为癌症早期诊断和治疗的靶标的巨大潜力,最近关于人类微生物组和癌症之间相关性的研究引起了各个科学领域的广泛关注,导致了受益于跨学科技术发展的显著进展。然而,在这一新兴领域仍然存在各种各样的挑战,例如肿瘤内细菌的低生物量和一些微生物物种的不可培养特性。由于肿瘤微生物组研究的复杂性(如肿瘤微环境的异质性),迫切需要具有高空间和时间分辨率的新方法。在这些发展中的方法中,多组学技术(基因组学、转录组学、蛋白质组学和代谢组学的组合)是强有力的方法,可以促进在中心法则的不同水平上理解肿瘤微生物组。因此,多组学(尤其是单细胞组学)将对未来微生物与肿瘤微环境相互作用的研究产生巨大影响。本文系统总结了多组学的研究进展及其在肿瘤微生物组研究中的现有和潜在应用,为研究者提供了一个可供参考的组学工具箱。
视频解读
Bilibili:https://www.bilibili.com/video/BV1YP4y1k7Md/
Youtube:https://youtu.be/FVNgyrAww6c
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
已知的将癌症和微生物联系起来的最早历史记录可以追溯到一个多世纪以前。目前地球上大约有1012种已知的微生物,只有11种被国际癌症研究机构(IARC)宣布为“致癌微生物”(或已知对人类致癌的病原体)。这些肿瘤微生物每年导致大约220万个病例(全世界大约13%的癌症病例)。此外,研究人员发现活微生物存在于多种肿瘤中(图1),包括乳腺癌,结肠直肠癌,肝细胞癌,胰腺癌,皮肤癌,肾细胞癌,胃癌,肺癌,前列腺癌,鼻咽癌等。值得注意的是,同类型肿瘤中的微生物有惊人的相似之处。已发现肿瘤内微生物通过多种机制影响肿瘤的发展和治疗,如DNA损伤、致癌途径激活、抗肿瘤药物分解代谢和免疫系统调节(图1)。
研究进展已经开始揭示肿瘤微生物群如何影响癌症的全部范围。这些微生物的一个公认的机制是释放能够诱导致癌DNA突变的基因毒素,包括来自大肠杆菌的大肠菌素(DNA烷基化剂)来自革兰氏阴性菌(如空肠弯曲杆菌)的细胞致死性扩张毒素(脱氧核糖核酸酶激活剂),以及作为产肠毒素脆弱拟杆菌的活性氧(ROS)生产者的毒素。此外,许多与癌症发病机制相关的调节途径会受到微生物的影响。例如,幽门螺杆菌,上述11种已公布的微生物之一,已被证明通过诱导炎症和影响调节粘膜细胞生长和增殖的关键细胞内信号通路而导致致癌。除了影响肿瘤发生之外,微生物群落还通过抗肿瘤药物的代谢参与癌症治疗方法。吉西他滨是一种常用于治疗胰腺导管腺癌的化疗药物,可被肿瘤内细菌代谢至失活状态,从而导致癌症耐药性。此外,许多微生物可以通过影响宿主免疫监视来影响癌症的发展和治疗。因此,肿瘤微生物组逐渐成为癌症早期诊断和治疗(尤其是免疫肿瘤学)的新焦点。
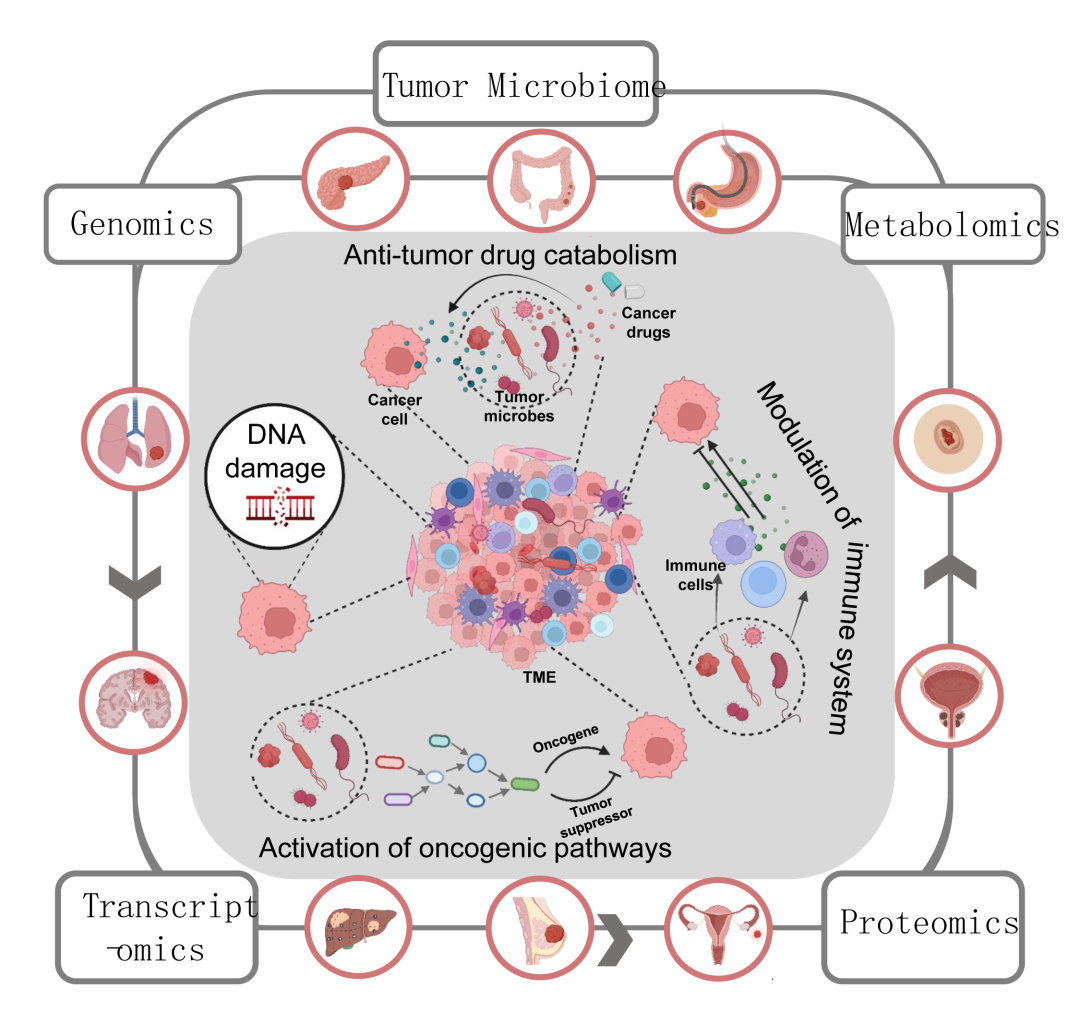
图1. 微生物的致癌作用以及多组学方法研究肿瘤微生物。TME,肿瘤微环境
尽管对肿瘤微生物组的研究已经取得了很大进展,但在这一新兴领域仍然存在许多挑战。我们对于肿瘤微生物组的了解还远远不够,这主要是来自于研究技术的限制。随着肿瘤微生物组在基础和转化研究中变得越来越重要,需要开发具有高空间和时间分辨率的新方法。最近,多组学技术(结合基因组学、转录组学、蛋白质组学和代谢组学)的综合应用在生物医学研究中显示出巨大的力量,并促进了对许多以前难以理解的生物学过程的理解。为了揭示肿瘤微生物组在癌症发展和不同水平的中心法则治疗中的病理生理学功能(图1),多组学(尤其是单细胞组学)方法已经开始被开发和应用。因此,在这篇综述中,我们将系统地总结可用于肿瘤微生物组研究的多组学技术并以甲基乙二醛为例介绍多组学在肿瘤微生物相关领域的研究。
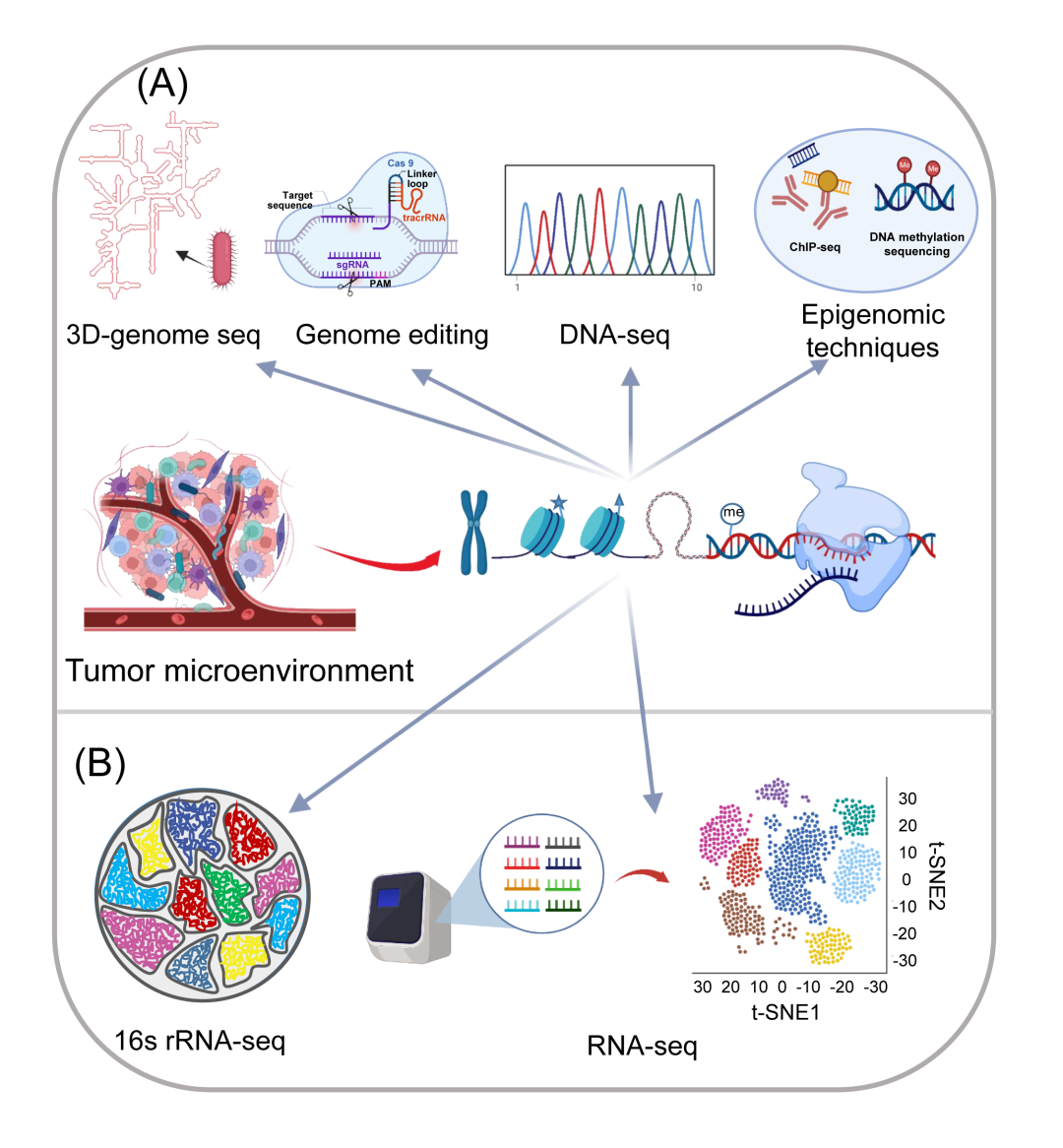
图2. 可用于肿瘤微环境研究的基因组和转录组研究技术
引用于肿瘤微生物组研究的基因组学和转录组学
基因组学和转录组学专注于核酸(即DNA和RNA),尤其是那些参与基因表达调控的核酸。目前,已经发展了大量的研究技术来研究不同生物系统的基因组和转录组。这些值得注意的技术包括高通量测序和基因编辑(图2)。目前,最广泛使用的测序技术包括三种二代测序(NGS)系统(即Illumina、Ion Torrent、BGI)和两种三代测序技术(即PacBio和纳米孔)。这些测序方法是发展各种基因组和转录组研究方法的基础,包括DNA测序,RNA测序,16S rRNA测序,表观遗传学技术(例如,ChIP-seq和DNA/RNA甲基化测序)和三维基因组技术。此外,基因编辑工具的最新研究进展已经能够在全基因组水平上进行多基因操作。由于这些技术可以很容易地靶向核酸,它们已经成为研究肿瘤微生物组和TME的重要工具,大大加快了肿瘤微生物的识别和可追溯性,并有助于揭示肿瘤微生物、癌细胞和免疫系统如何相互作用。
DNA测序
DNA-seq是微生物基因组研究中最重要的方法之一,可用于分析复杂样品中的所有基因,如人类微生物组。DNA-seq通常用于三种类型的基因组分析:全基因组测序(WGS)、全外显子测序(WES)和靶向测序。随着DNA技术的快速发展,DNA-seq正成为研究不可培养微生物(包括人类微生物组中的微生物)的有力工具。基因组文库的测序和异源表达极大地促进了我们对代谢途径和肿瘤微生物生理的理解。
16S rRNA测序
16S rRNA测序是一种专门针对细菌和古细菌表达的30S核糖体亚单位的16S组分的测序技术。该序列包含交替的高度保守和高变区。保守区可用于设计扩增靶片段的通用引物,而高变区的分析可用于鉴定特定的细菌物种,尽管一些注释可能低于物种水平。在肿瘤微生物组领域,16S rRNA测序主要用于研究群落中的物种组成、物种间的进化关系以及微生物群落的多样性。
RNA测序(RNA-seq)
RNA-seq是一种转录组测序方法,其中高通量测序方法用于研究复杂样品中不同RNA的群体,包括信使RNA(mRNA)、转移RNA(tRNA)、非编码RNA(ncRNA)、微小RNA(miRNA)等。在过去的几十年里,RNA-seq技术发展迅速,已经成为在转录组水平上分析基因表达不可或缺的工具。RNA-seq的应用范围随着NGS的发展而扩大。在RNA生物学领域,RNA-seq已被广泛应用于研究单细胞基因转录、蛋白质表达和RNA结构。RNA-seq技术的进步也促进了空间转录组学的出现和发展。直接长阅读或全长RNA-seq方法,以及更好的数据分析方法,使研究人员能够更好地了解RNA生物学,包括转录过程的机制以及折叠和分子间相互作用对RNA功能的影响。总的来说,RNA-seq是一个强有力的工具,通过在基因转录水平上分析来研究癌细胞和肿瘤微生物组对彼此通路的相互影响。
表观遗传学技术
表观遗传学是指不涉及DNA序列改变的表型变化的研究。与遗传调节不同,典型的表观遗传变化(包括DNA/RNA修饰和组蛋白翻译后修饰)是可逆的,并且不影响细胞基因组序列。在真核细胞中,核小体是染色质组织的基本重复单位,由围绕核心组蛋白八聚体(即H2A、H2B、H3和H4的两个拷贝)缠绕1.5次的147个碱基对组成。DNA、RNA和组蛋白共价修饰的动力学是控制特定基因激活和抑制的表观遗传调控的关键机制。表观遗传调控在复制、转录和DNA损伤修复过程中对染色质结构和功能的调节起着重要作用,与许多人类疾病特别是癌症密切相关。
具体来说,DNA甲基化改变了基因转录而不改变基因组序列,这可能导致肿瘤发生,当肿瘤抑制基因被关闭时。大量研究表明,DNA异常甲基化与多种肿瘤的发生发展密切相关。因此,DNA甲基化水平的变化已被检测为癌症诊断的生物标志物。目前,已经开发了多种DNA甲基化测序技术,例如全基因组亚硫酸氢盐测序(WGBS),简化的亚硫酸氢盐测序(RRBS),氧化亚硫酸氢盐测序(oxBS-seq),以及无细胞甲基化DNA免疫沉淀和高通量测序(cfMeDIP-seq)。
另一方面,组蛋白翻译后修饰(PTMs)包括甲基化、磷酸化、乙酰化、巴豆酰化、泛素化、糖基化、糖化和ADP核糖基化。各种组蛋白PTMs被认为与癌症的发生和发展有关。例如,来源于微生物的甲基乙二醛合酶(MGSs)可以在TME中生物合成过量的甲基乙二醛。组蛋白MGO-糖基化已被证明通过影响染色质的三维结构来影响肿瘤的发展。
染色质免疫沉淀测序(ChIP-seq)是研究组蛋白修饰对基因转录影响的最常用方法之一,尽管它最初是为了研究蛋白质和DNA之间的相互作用而开发的。染色质免疫沉淀(ChIP),也称为结合位点分析,是研究体内这些相互作用的有力工具,对于更好地理解基因组的表观遗传变化至关重要。ChIP-seq结合了ChIP和第二代DNA测序技术,可以有效地检测特定组蛋白修饰和转录因子的DNA结合位点。ChIP-seq的原理如下:在纯化之前,通过ChIP富集和免疫沉淀与感兴趣的蛋白质结合的DNA片段,并构建文库。然后对富集的DNA片段进行高通量测序。通过应用这种方法,研究人员已经精确定位了基因组中与特定组蛋白相互作用的序列和转录因子。对于肿瘤微生物组研究来说重要的是,ChIP-seq适用于宿主和微生物细胞的基因组研究。
三维基因组学
三维基因组学,也称为空间基因组学,基于一维(1D)基因组序列的基本信息,研究基因组的三维结构,以及不同元件(包括转录因子和DNA/RNA结合蛋白)介导的转录调控机制。近年来,在该领域发展的技术主要包括Hi-C(高通量/高分辨率染色体构象捕获),Micro-C(基于微球菌核酸酶的染色体折叠分析),Micro-C XL(使用长x-接头的基于微球菌核酸酶的染色体折叠分析),ChIA-PET(通过成对末端标签测序的染色质相互作用分析)和HiChIP(原位Hi-C,随后是染色质免疫沉淀)。与肿瘤微生物组研究相关,虽然上述这些方法大多集中在宿主细胞的真核基因组上,但Hi-C技术最近已成为研究细菌染色体的有力工具,能够分析样品中DNA序列的物理邻近性。
基因组编辑
基因编辑(也称为遗传修饰或基因工程)主要是指使用生物化学的方法来修改基因组序列。这通常通过将靶基因片段引入基因组或从基因组中删除特定基因区域来实现,从而改变宿主细胞的基因型或增强原始基因型。由于基因编辑有着悠久的历史,许多优雅的工具被开发为“基因手术刀”,包括人工限制酶锌指核酸酶转录激活因子样效应核酸酶(TALENs),聚集的有规则间隔的短回文重复序列(CRISPR)和CRISPR相关蛋白9(Cas9),CRISPR基本编辑器,主要编辑系统以及通过位点特异性靶向元件(PASTE)的可编程添加。目前,CRISPR-Cas9技术及其衍生的系统是基因组编辑中应用最广泛的主流技术。此外,Cas9核酸内切酶dead(也称为dead Cas9或dCas9)是Cas9的突变形式,由于其核酸内切酶结构域中的点突变,其不能再作为核酸内切酶。dCas9作为基因定位向导,通过与其他功能酶(如胞苷脱氨酶)的融合表达,现已广泛用于不同生物系统的基因组编辑,腺嘌呤脱氨酶、以及表观遗传的书写/擦除酶和其他由此产生的新兴技术(如表观基因组编辑)。
至于肿瘤微生物组研究,基因组编辑已广泛应用于研究两种宿主细胞和微生物。例如,据报道,肠道微生物的基因组组成变化很大。单核苷酸多态性(SNPs)的系统发育分析显示了全球宏基因组样本中显著的种内遗传变异。微生物菌株的基因组存在相当大的差异,同一物种可能使用相似的序列来实现不同的功能。因此,为了理解基因组变异的作用,用CRISPR-Cas9编辑是修饰微生物分离物的有前途的方法。
单细胞基因组学和转录组学
基因组和转录组方法可以实现更高的覆盖率和更低的伪影风险,但它们的组装取决于测序深度和社区复杂性。例如,宏基因组方法允许研究人员研究复杂群落中的细菌,但这些群落的复杂性意味着低丰度细菌的序列可能被高丰度生物的序列抑制,或者在人类微生物组的情况下,被线粒体DNA抑制。此外,具有相似表型的细胞中的遗传信息可能显著不同,并且大量的低丰度信息可能在整体表征中丢失。为了弥补这些局限性,单细胞测序技术旨在揭示单个细胞的基因结构和基因表达,更好地反映细胞间的异质性。
查看目前使用单细胞技术分析肿瘤微生物群的出版物,有四种主要的研究方法来研究基因组和转录组:琼脂平板稀释基于精细毛细管移液分离系统和倒置显微镜的单细胞外显子组测序基于液滴的微流体,以及改进的荧光原位杂交(FISH)。值得注意的是,分析单细胞的第一步和关键步骤是从组织或其他来源制备单细胞悬液。
琼脂平板稀释法
琼脂平板稀释是指通过平板划线获得单一微生物的纯培养物的方法,并且是获得微生物纯培养物的最传统的方法。通过反复稀释微生物或不同细胞类型的混合物,人们可以将不同的微生物菌落分离成单细胞,这些单细胞将生长成同质菌落(图3A)。微流控划线平板(MSP)技术的发展建立在使用微流控技术的传统平板划线的基础上。MSP使用微流体制造微液滴,可以在培养皿上划线培养单细胞。传统的琼脂平板稀释和MSP都可以整合到高通量筛选工作流程中。
基于倒置显微镜和口控毛细管移液系统方法
来自组织的连续稀释的细胞悬浮液可以在倒置显微镜下使用口控毛细管系统或其他单细胞移液系统进行处理并分离成单细胞。将分离的单细胞随机分离到PCR管中,并在用显微镜进行视觉确认后记录为显微照片。然后可以提取单细胞基因组用于全基因组扩增(WGA)。如前所述,该扩增的基因组可用于外显子组测序(图3A)。这种方法已被用于研究肾细胞癌和邻近肾组织的肿瘤内遗传景观,发现这种癌可能比以前认为的更复杂,并促进了更有效的细胞靶向治疗的使用。值得注意的是,这种方法也可用于研究单个肿瘤细胞内的微生物。
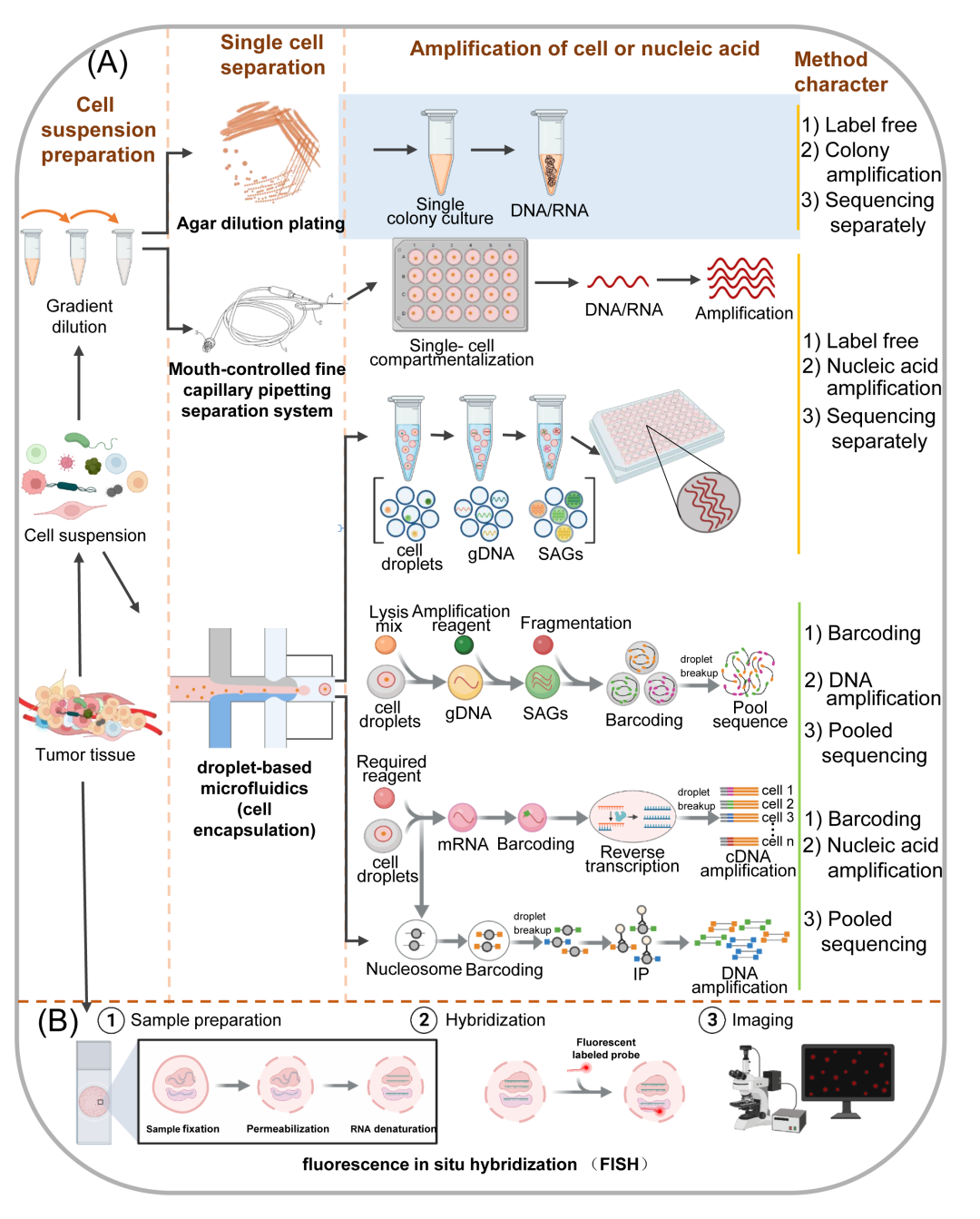
图3. 单细胞基因组和转录组方法
(A)从肿瘤组织制备混合细胞悬液(左),将混合物分离成单细胞以进行基因组和转录组测序的三种方法的工作流程(中),琼脂稀释平板、口控毛细管移液分离系统、基于液滴的微流控技术(细胞封装),gDNA,指导DNA单细胞扩增基因组,每种方法的特点比较(右);(B)单细胞RNA荧光原位杂交的工作流程。
微滴微流体法
这种微流体方法主要用于通过使用两种不相容的液体来制备液滴,液滴的产生由微管结构和两种液体之间的流速比来控制(图3A)。由固定流速的泵驱动,两种液体进入不同的微通道,当它们相遇时,一种流体剪切另一种流体,产生微滴。这些液滴是理想的微反应室,其大小可以容纳单个细胞。使用这些系统可以对单个液滴进行填充、操作、分离、组合、检测和分类,并且使用微升试剂可以操作数千个单个液滴。微滴微流体可以将大量群体分成单个细胞,通过一系列酶促步骤提取和分割基因组。在这些单细胞被测序之前,遗传物质被放大和标记。这是一项结合了微流体技术、DNA条形码和测序技术的综合技术。
在裂解和纯化基因组DNA之前,被包封的微生物可以直接在液滴中培养,并在第一次基因组扩增过程中生长至数百个细胞。然后,使用细胞分选仪将含有单细胞扩增DNA的珠子分选到标准PCR板中,随后再扩增到单细胞扩增基因组(SAG)文库中。微流控液滴发生器已用于在琼脂糖凝胶珠中培养小鼠肠道微生物,这是一种基于SAG-gel平台的典型单细胞测序方法(图3A)。
被包封的单一微生物也可以直接裂解,然后进行DNA扩增或RNA逆转录和条形码标记。对于单细胞基因组测序,细胞通常首先被扩增,然后用条形码标记。对于单细胞转录组测序,通常首先标记细胞RNA,然后是逆转录,最后是互补DNA(cDNA)扩增(图3A)。为了研究细胞内的核小体,核小体必须首先用条形码标记,然后进行免疫沉淀(单细胞芯片-seq)和DNA扩增(图3A)。微滴微流体技术可以分析数百万个独立的反应。最近,它被用于单个DNA分子的深度测序,核小体被标记以通过单细胞芯片测序和高通量分析进行分析。
无需单独培养微生物即可对细胞进行测序的能力是微滴微流控单细胞测序的一个重要方面。通过这种方法,可以生成宏基因组数据库来研究肿瘤和其他区域(如肠道)中的微生物。这种方法的局限性在于,它从单个基因组拷贝开始,并且在酶和微流体处理过程中材料的损失是不可挽回的,这限制了最大可用覆盖范围,并导致每个细胞的覆盖范围显著降低。
改进的荧光原位杂交法
荧光原位杂交(FISH)一直是研究培养微生物的重要工具,可以直接用于鉴定单个细菌细胞,尽管传统的FISH受到其低分辨率的限制。FISH包括使用单链寡核苷酸探针来特异性标记靶RNA(图3B)。然而,因为荧光的程度与靶RNA的拷贝数直接相关,所以这种技术最适合于靶向rRNA。由于16S rRNA的不同区域具有不同的保守水平,探针可以是物种特异性的或根据不同的分类水平进行选择。为了克服低分辨率,研究人员开发了各种方法,包括催化报告分子沉积-荧光原位杂交(CARD-FISH),也称为酪胺信号放大(TSA)FISH,它使用辣根过氧化物酶(HRP)标记的探针以指数方式放大信号,以便准确和特异性地对单细胞进行荧光标记。高度系统发育分辨率FISH(HiPR-FISH)是另一种新设计的方法,它修改了单细胞自动图像分割中的现有算法,并执行像素分类和图像优化滤波。因此,这些进展使研究人员能够更好地研究特定的微生物种群,可视化和分类少量的细菌和非常低拷贝的RNA,并实现单细胞定量。很可能在使用其他分析方法对肿瘤组织进行分类和研究之前,FISH将首先用于识别和定位肿瘤组织中的微生物。
蛋白质组学
基因组和转录组的表达和调节直接反映了成千上万的蛋白质,其特征,包括表达水平、PTMs和蛋白质-蛋白质相互作用,是错综复杂的。质谱与液相色谱(LC-MS)联用是此类分析中最常用的方法之一并且已经成为肿瘤微生物组研究的有力工具。蛋白质组分析有三种不同的策略:“自上而下”、“中间向下”和“自下而上”。具体来说,“自上而下”(图4A)直接将目标蛋白质送至质谱(MS)进行分析,无需任何预处理。“中部向下”指的是蛋白质被部分消化以获得相对较大的肽段,然后被送到质谱进行进一步分析。最后,“自下而上”分析,也称为鸟枪法分析,是肽段分析最长使用的方法,在质谱分析之前,将蛋白质消化至6-20个氨基酸(图4B)。在这三种策略中,第三种策略是全球蛋白质组研究人员最广泛使用的,因为自下而上的分析策略通常对于具有高复杂性的蛋白质混合物更有效,这种蛋白质混合物可以包含数千种不同数量级的混合蛋白质。
在蛋白质组学中,一个主要目的是比较感兴趣的样品(如健康和患病组织)以识别差异表达的蛋白质并量化这些差异。目前有两种产生鸟枪法MS蛋白质组学数据的主要方法:数据依赖采集(DDA)和数据独立采集(DIA)(图4B)。在串联质谱(MS/MS)中,DDA策略仅选择在MS的第一个循环中产生的信号较高的某些肽段在第二个循环中进行断裂,而使用DIA方法,在第一个MS循环中产生的所有肽段都可以在第二轮中断裂。
DIA已成为利用临床样本(如肠道微生物样本)进行致病机制和生物标志物筛选研究的最受欢迎的选择和肿瘤因为它可以获得对样品蛋白质组的无偏见和更深入的观察,特别是当这些样品来自研究不足的生物体(例如,肿瘤微生物)或复杂组织(例如,肿瘤组织)时。与DIA方法相比,DDA是目标分析中更广泛应用的定量方法。靶向蛋白质组学分析主要通过平行反应监测(PRM)来关注样品中感兴趣的蛋白质子集或多重反应监测(MRM)。MRM,也称为选择性反应监测(SRM),是一种高度特异和灵敏的MS技术,可以选择性地定量复杂混合物中的肽。该技术使用三重四极杆质谱(TQMS,QqQ),首先靶向与目标蛋白质对应的肽离子,随后将其裂解以产生一系列子离子。可以选择这些分裂的子离子中的一个(或几个)用于定量目的。PRM与SRM/MRM不相上下,但更便于分析开发,因为它基于高分辨率和高精度质谱对蛋白质和肽进行绝对定量。蛋白质定量方法一般包括绝对定量和相对定量。在建立标准曲线后,绝对定量确定样品中靶肽的表达水平,对于该标准曲线,标准肽的精确量是已知的。相对定量通过比较强度来确定两个样品之间表达的倍数变化,而不需要获得标准曲线。
相对定量法有标记法和无标记法。无标记定量方法旨在确定两种或多种生物样品中蛋白质的相对量,而不使用与蛋白质结合的稳定同位素标记。常用的标记定量技术包括相对和绝对定量的同量异位素标记(iTRAQ)串联质量标签(TMT),以及在细胞培养物中通过/用氨基酸进行稳定同位素标记(SILAC),并通过用同位素标记物标记或取代相应的氨基酸来实现。TMT(图4D)和iTRAQ用于体外标记,而SILAC是一种体内标记技术(图4C)。这些标记方法的优点是可以在单个实验中测定不同来源的蛋白质的量。SILAC策略突出表现为用同位素重形式的氨基酸替代天然存在的轻形式的氨基酸。由于在样品制备的初始步骤中结合了标记和未标记的样品,SILAC最大限度地减少了平行处理样品可能导致的定量误差。此外,样品的混合允许多种富集技术,包括免疫沉淀。这些技术可以改善对低丰度蛋白质和PTMs的丰度变化的检测,例如磷酸化和糖基化。
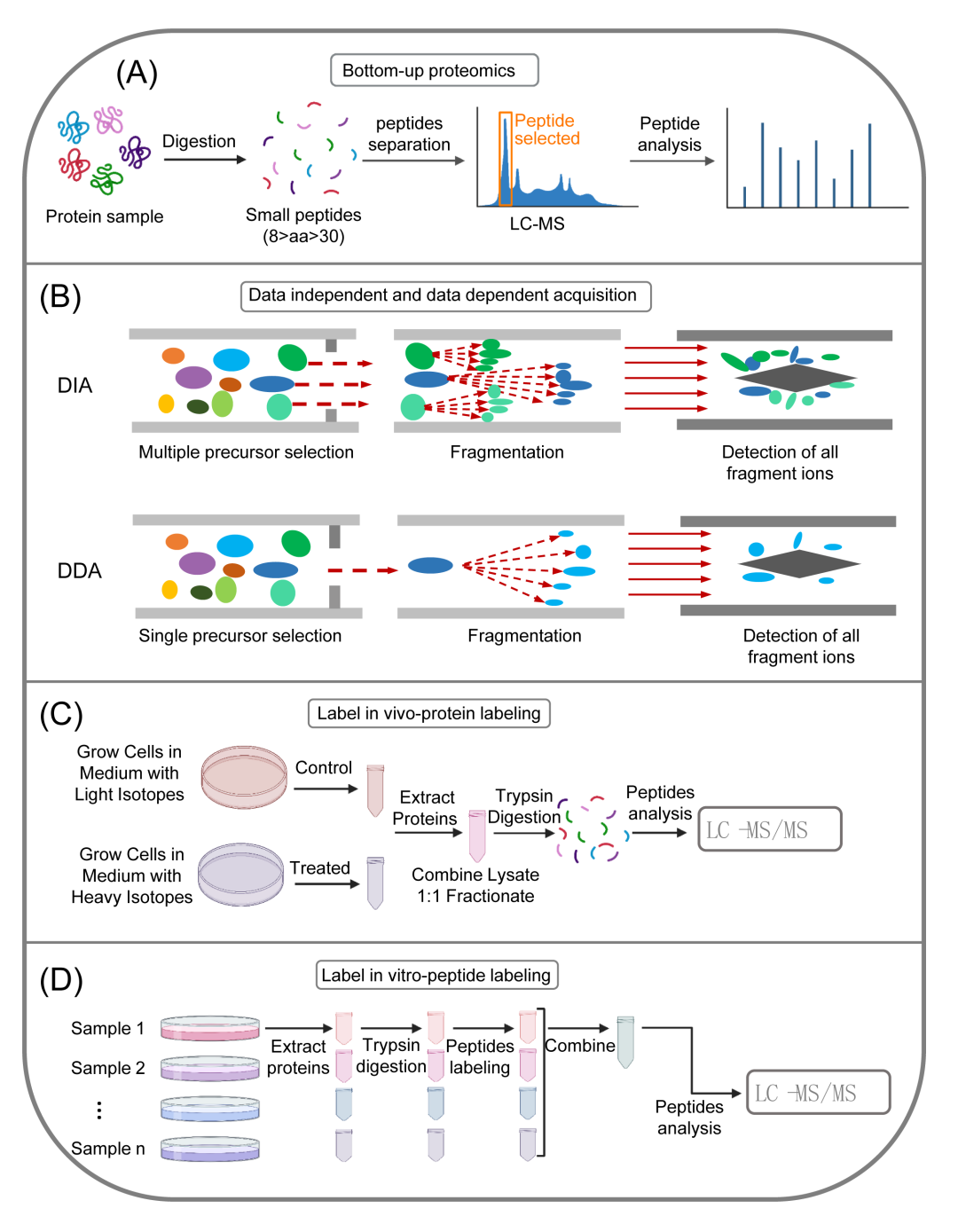
图4. 蛋白质组学的不同技术
(A)自下而上蛋白质组学的程序;(B)蛋白质组学的数据独立(DIA)和数据依赖获取(DDA)模式;(C)使用SILAC(细胞培养中由/用氨基酸进行稳定同位素标记)的工作流程,这是一种体内标记定量蛋白质组学的方法;(D)使用TMT(串联质量标签)的工作流程,TMT是一种体外标记定量蛋白质组学方法。
代谢组学
代谢组学广泛应用于定量分析不同生物体内的所有代谢物,并发现这些代谢物相应的病理生理功能。代谢组学在肿瘤微生物组研究中的应用致力于识别肿瘤微生物的代谢差异及其与周围环境的相互作用,增进对肿瘤微生物如何影响致癌相关分子机制的理解。这些代谢物主要包括短链脂肪酸,氨基酸代谢物,维生素,胆汁酸,乳酸,毒素和甲酸盐,等等。许多研究表明,微生物代谢产物可以通过正向或负向调节(图5A),通过影响免疫、炎症、信号通路,或通过调节肿瘤微生物,显著影响肿瘤的发生和发展。
例如,SCFAs是肠道菌群的主要代谢物之一。研究表明,SCFAs可以对肠道功能和肠道代谢产生积极和消极的影响。已经发现补充某些产生SCFA的细菌可以抑制肠道肿瘤的发展。另一方面,SCFAs也有望成为结直肠癌的诊断和治疗靶点。异戊酸(IVA),一种与结直肠癌相关的肠道微生物的SCFA代谢物,可以促进蛋白质的表达,导致下游5-羟色胺(5-HT)的增加。5-HT直接作用于肿瘤干细胞,促进自我更新,增加肠道肿瘤发生。
产生维生素B6和B9的细菌的中断降低了最常见的化疗药物之一5-氟尿嘧啶(5-FU)的疗效。此外,肠道细菌分泌的维生素E可以进入树突状细胞(DCs),增强DCs的活化,促进T细胞抗肿瘤免疫反应。
微生物的其他代谢产物也可以调节T细胞的代谢,如次生胆汁酸(BA)异石胆酸,可以增强氧化磷酸化和线粒体ROS的产生,促进调节性T细胞(Treg)的分化。一些反流的BA和肠道微生物之间的相互作用可导致次级BA牛脱氧胆酸的产生,其促进胃癌的发生。产毒素细菌艰难梭菌可激活肠上皮祖细胞中的信号通路,导致ROS产生增加和促进肿瘤的粘膜免疫反应,包括活化髓样细胞的浸润和产生白细胞介素-17(IL-17)的淋巴细胞亚群的增加。具核梭杆菌分泌的甲酸盐可以激活许多信号通路,增加结直肠癌细胞的侵袭力。来自肠道菌群的氧化三甲胺(TMAO)增强CD8+ T细胞介导的抗肿瘤免疫。没食子酸是肠道微生物的另一种代谢产物,已被证明可以调节Treg细胞并增强癌症免疫治疗,同时在p53突变存在的情况下也导致细胞中的恶性表型。肠道细菌及其代谢产物可通过诱导突变、增强促瘤炎症反应、招募免疫抑制细胞等方式促瘤。肠道真菌还可以诱导促瘤炎症微环境,并可以产生真菌毒素诱导突变,从而促进肿瘤。
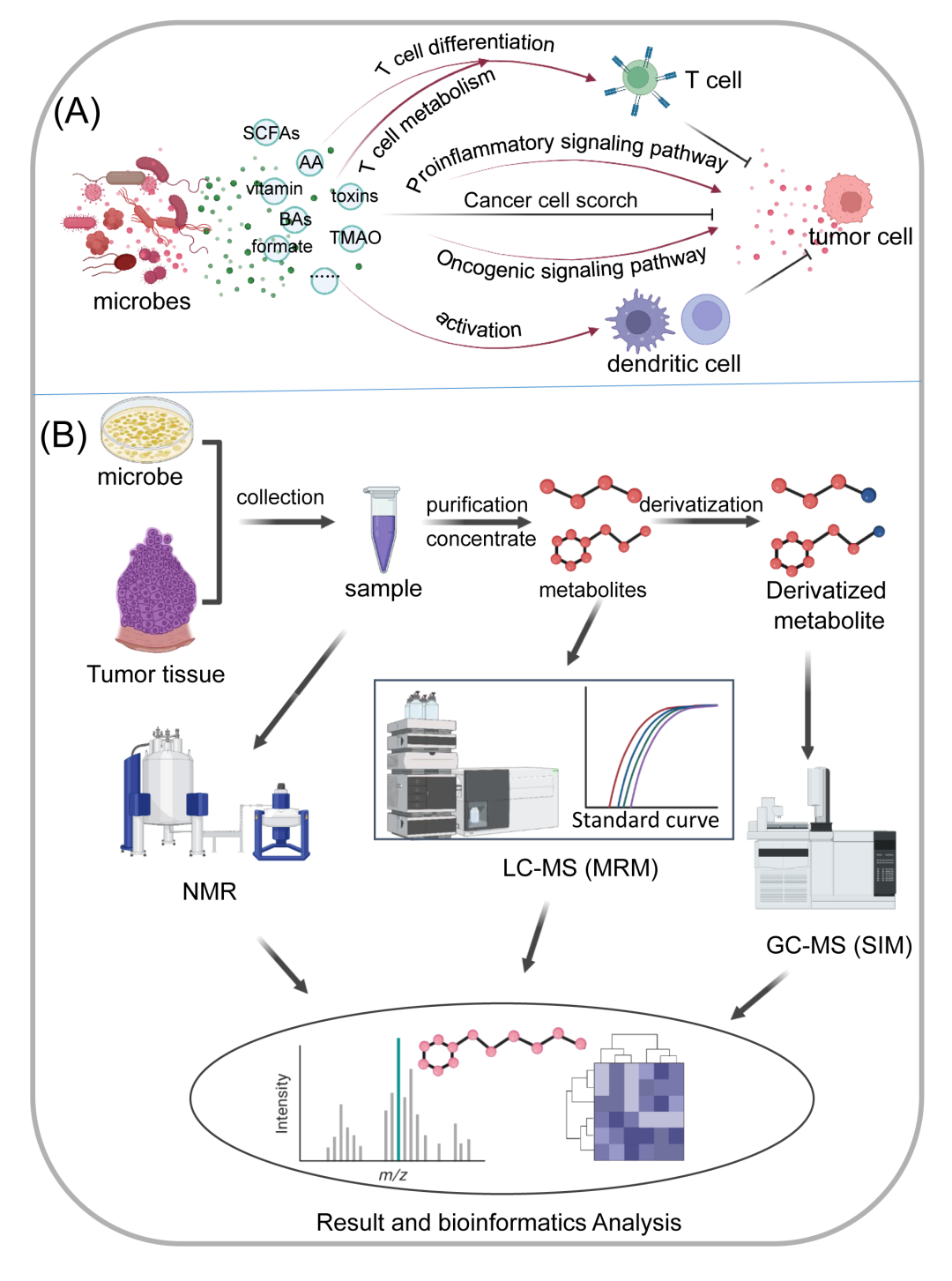
图5. 用于肿瘤微生物组研究的代谢方法
(A)微生物代谢物通过多种机制影响肿瘤;(B)三种最常用的肿瘤微生物代谢组学研究方法:核磁共振、多反应监测液相色谱质谱和选择离子监测气相色谱质谱。
代谢组学依赖于分析技术的使用。两个仪器平台(图5B),核磁共振(NMR)和MS,都是目前的首选。NMR是非破坏性的,不依赖于分析分离技术,样品制备相对较快,易于操作,成本较低。通过向样品中加入已知浓度的内标化合物,也可以提高使用NMR的绝对定量。通过NMR进行分子表征的原理在于其强磁场,该强磁场与分子的核相互作用,因为它们吸收并共振特定频率的电磁光谱。然而,由于核磁共振的灵敏度相对较低、微生物代谢物的复杂性以及代谢物浓度的变化,核磁共振在微生物代谢组学中的应用受到了限制。
为了克服这些问题,高分辨率质谱成为代谢组学研究中不可或缺的工具。该技术利用各种分离技术,包括液相色谱(LC)、气相色谱(GC)和毛细管电泳(CE),并广泛用于微生物代谢组学。气相色谱-质谱联用技术是代谢组学中最常用的技术之一,因为它性能稳定,结果重现性好,峰容量高。该技术主要用于挥发性和热稳定性低分子量化合物。然而,大多数微生物代谢物,如磷酸化代谢物,是非挥发性的和热不稳定的,因此,当放置在较高温度下时可能会降解。因此,这种方法通常用于分析衍生的微生物代谢物,如来自费氏丙酸杆菌、大肠杆菌和枯草芽孢杆菌的代谢物。Miyamoto等人还利用GC-TOF-MS发现了与肺癌预后相关的生物标志物。CE-MS是代谢组学分析的另一个工具。与GC-MS和LC-MS相比,它的优点包括分离效率高、样品体积小、成本低。但是它的主要缺点是毛细管电泳(CE)和MS的接口比较困难。
所以CE-MS没有得到广泛的应用。LC-MS和串联LC-MS/MS是代谢组学中重要的分析工具。与GC-MS不同,LC-MS可以分析极性和高分子量化合物,无需高温或挥发性,具有更广泛的代谢物分析范围,并且样品制备更方便。此外,LC-MS具有更高的灵敏度,即使样品数量很少。超高效液相色谱(UHPLC)的出现大大提高了LC-MS的色谱分辨率
代谢组学研究通常分为两类:靶向代谢组学和非靶向代谢组学。非靶向是指系统分析样品中的所有代谢物,而靶向是指代谢变化的相对或绝对定量,绝对定量通常使用SRM和建立的标准曲线。TQMS和传统的离子阱质谱仪已经广泛用于靶向代谢组学研究。然而,非靶向代谢组学的实施需要更先进的分析方法、自动化光谱数据处理、生物数据解释和假设生成。高分辨率串联质谱仪,如四极杆飞行时间(Q-TOF)质谱仪、傅立叶变换离子回旋共振(FT-ICR)质谱仪和基于离子阱质量分析器轨道阱的质谱仪,是非靶向代谢组学研究的优选仪器,因为它们可以阐明结构信息并允许高数据采集速度。
细胞内代谢可以根据时间和外部环境快速变化。因此,取样和样品制备的方法和条件,包括时间、储存条件和其他因素,会极大地影响代谢检测的再现性、精密度和准确度。样品制备分为两个阶段:代谢淬灭和代谢物提取。淬灭是在有限细胞膜损伤的情况下快速停止细胞代谢活动的过程。快速收集组织或细胞,立即冷冻在液氮中,并储存在-80℃。此外,也常用其他方法,如冷冻淬火或化学淬火,用酸或冷的含水甲醇溶液。
提取是从样品中分离代谢物的过程。提取可以通过物理方法实现,通过手工研磨或使用组织匀浆器。使用有机溶剂(如甲醇、氯仿和乙腈)进行溶剂萃取是更常用的方法,因为它具有更高的萃取效率和LC-MS分析中最小的电离抑制。基于NMR的分析不需要化学衍生,样品制备也简单得多。这两种微生物代谢分析方法各有优缺点。研究人员应根据感兴趣化合物的特性和分析工具的可用性,选择最佳方法或工具组合来分析微生物代谢物。
单细胞蛋白质组学和代谢组学
单细胞测序用于分析细胞基因组、转录组和表观基因组。同样,单细胞蛋白质组和代谢组研究对于单细胞水平的基因组和转录组研究也是必不可少的。与单细胞核酸测序不同,在单细胞核酸测序中信号可以被放大,单细胞蛋白质和代谢物分析信号不能被放大。在这类研究中,追踪是最大的挑战,因为这对于减少样品处理过程中细胞内分析物的损失至关重要。
单细胞蛋白质组学领域仍处于起步阶段,缺乏明确和普遍适用的方法。尽管如此,最近在单细胞蛋白质的绝对定量和高度多重蛋白质测量方面的快速进展依赖于MS仪器灵敏度的提高和样品制备方法的改进,以克服单细胞中痕量蛋白质的局限性。
在连续几代质谱仪中,基于Orbitrap的高分辨率MS仪器在离子传输效率、检测器灵敏度、占空比和分辨率方面经历了持续的改进。最近,当在最新一代的Orbitrap Eclipse MS上分析单个HeLa细胞时,肽和蛋白质覆盖率分别增加了36%和20%。最新的质谱仪器还引入了基于漂移管的离子迁移谱(DT-IMS),捕获离子迁移谱(TIMS)和高场不对称波形离子迁移谱(FAIMS),它可以提高选择性,从多电荷肽中分离或过滤单电荷离子,并选择多电荷肽进行自我测定。就鸟枪蛋白质组学的鉴定和定量而言,去除单电荷物质有利于痕量样品分析,因为在批量研究中可以忽略的溶剂簇和污染物在低投入研究中会变得非常重要。在所有情况下,单细胞分析中的LC分离和MS采集设置(如色谱柱、流速、离子注入时间和自动增益控制(AGC)目标设置往往与批量研究中使用的设置大相径庭,从而确保有足够的报告离子信号可用于准确定量和最大化离子利用率。然而,目前的单细胞代谢组学技术主要利用培养微生物的单细胞分析,不能直接应用于多细胞生物以研究微生物-细胞相互作用。质谱成像(MSI)通过绘制组织中的代谢物和其他生物分子,在单细胞代谢领域显示出巨大的潜力。
基质辅助激光解吸电离(MALDI)质谱是一种MSI技术,是目前另一种最常用的单细胞代谢分析技术。将样品与基质混合并用脉冲激光照射。在将分析物发送到质谱仪之前,光束使分析物质子化或去质子化。MALDI-MS测量含有平板细胞的组织切片或载玻片上不同点的质谱,使研究人员能够创建代谢的空间图。使用MALDI获得了空间分辨率低于5微米的质谱图像,这使得研究代谢物的分子分布更加可行。与其他方法相比,MALDI-MS因需要更少的样品制备和更高的通量而脱颖而出,并已成功用于揭示单细胞生物体克隆群体中的异质性和发现组织中的特化微生物和细胞亚型。MALDI成像的两个最大限制是低代谢物覆盖率和低电离效率。为了克服低代谢物覆盖率,芬斯特拉等人提出了一种MSI方法,该方法使用具有不同电离度的多种基质来靶向双极性,以及串联MSI,尽管低电离度的代谢物仍然有限。电离效率可以通过组织上的化学衍生来提高。开发了更新更灵敏的MALDI-2,通过在第二个激光器产生的气相激光羽流中启动额外的电离过程,增强了分析物信号。
这些进展表明,对MSI的改进可以增加我们对细胞和亚细胞水平上不同代谢物的了解。作为一项不断发展的技术,MSI仍有许多挑战需要克服。从MSI获得的数据很复杂,可能包含来自已知和未知代谢物的信号。软件和数据库的改进对于高度可靠地鉴定代谢物并得出准确的生物学结论至关重要。此外,定量方法的进步对于区分可能受基质效应或分析物电离效率影响的结果至关重要。
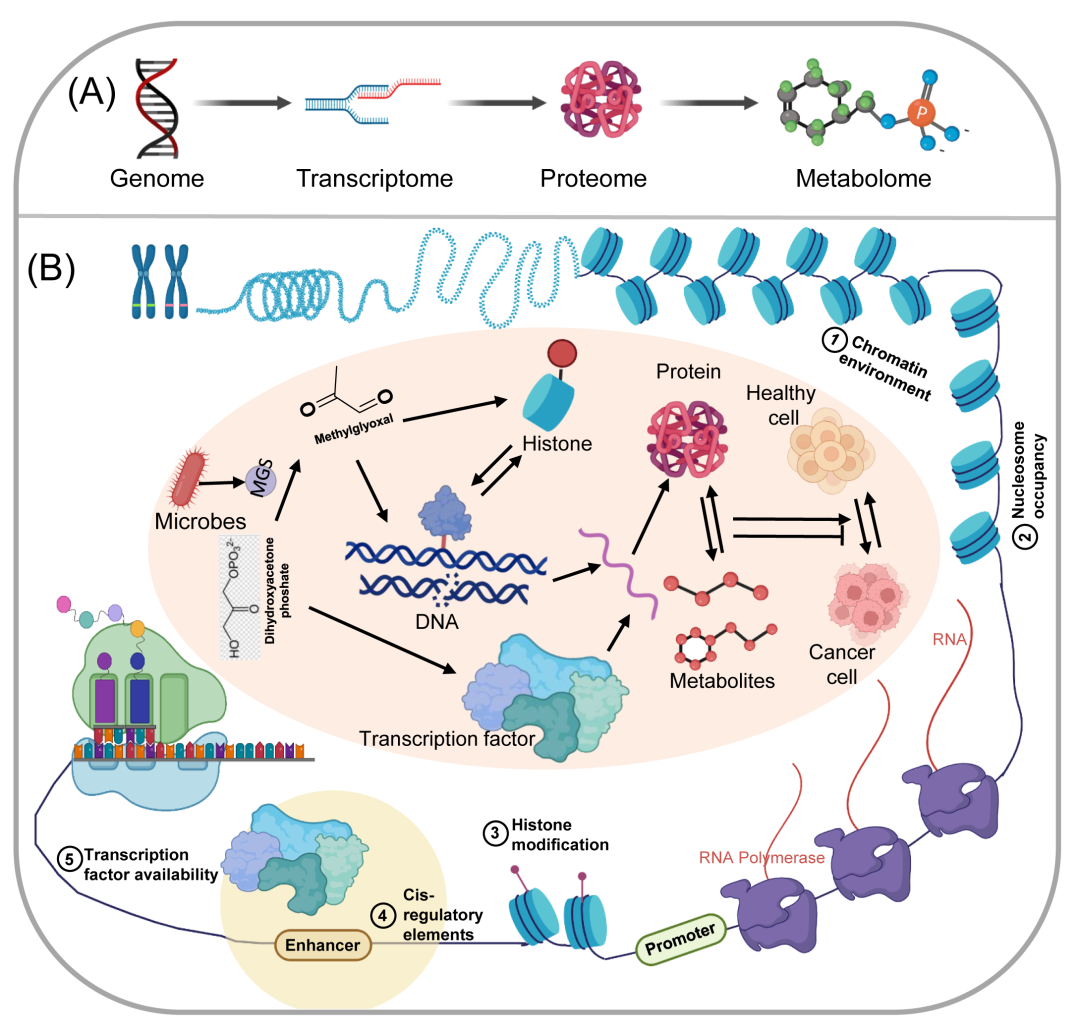
图6. 多组学应用促进相互关联学科的综合研究
(A)组学技术的流动,通过中心法则的各层,可以组合成多组学;(B)代谢物甲基乙二醛可以影响多组学网络的许多水平,包括基因、蛋白质、组蛋白和染色质。所有这些途径都有助于肿瘤的发展,MGS,甲基乙二醛合酶。
应用多组学方法进行肿瘤微生物组研究
我们越来越意识到,单一的组学领域不足以全面理解更复杂的生物学过程,如与肿瘤微生物组相关的过程。综合多组学试图填补这一研究空白,是探索这些主题的一个潜在的新方向。
已有研究表明,微生物产生的多种细胞毒性代谢产物在肿瘤发生和发展中起重要作用。使用代谢物甲基乙二醛(MGO)作为案例研究,已知MGO以各种方式影响人类健康。氧化镁,有氧和无氧糖酵解的非酶副产品和由MGSs酶促合成的微生物代谢物,可以共价修饰蛋白质而无需酶,并调节细胞功能,包括代谢和转录。据报道,MGO可以通过与鸟嘌呤残基反应来修饰DNA和RNA,诱导DNA和RNA损伤,并通过在有丝分裂期间破坏染色体分离来直接影响基因组的完整性。此外,MGO可以与蛋白质的赖氨酸残基反应,导致高级糖基化终产物(AGEs)的形成,进而导致细胞功能障碍和炎症。MGO是体内最有效的糖化剂之一,可糖化组蛋白的赖氨酸和精氨酸残基,这可导致代谢紊乱、表观遗传改变和核小体不稳定。据报道,MGO可以影响转录因子的活性并改变基因转录,如通过RNA测序所见,并且可以导致细胞应激因子的积累。组蛋白MGO糖基化的两阶段模型表明,低量的MGO通过促进复杂的转录有利于癌细胞的增殖,而过量的MGO导致染色质交联,削弱转录并导致细胞死亡。
化学蛋白质组学方法表明,高浓度的MGO可以诱导染色质中的组蛋白-组蛋白和组蛋白-DNA交联,这可以损害其动力学性质。合成和应用化学探针研究MGO糖基化可以提高追踪、丰富和研究MGO糖基化的重要工具的可及性和可行性,并促进对其潜在生化功能的理解。MGO在基因组、转录组、蛋白质组和代谢组水平上调节肿瘤的发生和发展(图6)。对MGO的研究表明,使用单一组学方法不足以全面了解这些蛋白质在生物学研究中的作用,并提出了其他代谢物和蛋白质的案例研究,可能需要更多跨学科的多组学方法。
未来展望
肿瘤微生物在肿瘤的发生、发展和治疗中起着重要的作用。微生物、肿瘤细胞和免疫细胞之间的相互作用可以极大地影响TME。免疫细胞不仅影响微生物与抗癌疗法的关系而且还可以直接调节微生物对癌症发展的影响。基于此,将微生物治疗与免疫治疗相结合成为癌症治疗研究的有效方向。多组学技术已经被应用于鉴定由TME中的肿瘤细胞和微生物产生的癌症生物标志物,从而极大地促进了新的诊断和治疗策略的发展。尽管通过组学方法在该领域取得了许多进展,但该领域仍处于起步阶段。许多研究技术还不成熟,例如单细胞技术在肿瘤微生物蛋白质组学和代谢组学研究中的应用,在很大程度上仍然是理论性的。此外,肿瘤微生物组的多组学研究缺乏统一的标准流程。总之,本文系统总结了可用于肿瘤微生物组研究的多组学方法及其优缺点。由于肿瘤内微生物的低生物量和TME的异质性,我们认为单细胞多组学将成为肿瘤微生物组研究中最有力的工具,尽管在开发相应的方法学方面还有很长的路要走。
引文格式:
Nan Zhang, Shruthi Kandalai, Xiaozhuang Zhou, Farzana Hossain, and Qingfei Zheng . 2022. “Applying multi‐omics toward tumor microbiome research.” iMeta. e73. https://doi.org/10.1002/imt2.73
作者简介

张楠(第一作者)
● 俄亥俄州立大学詹姆斯综合癌症中心癌症代谢中心博士后,毕业于北京大学基础医学院
● 目前研究方向为化学蛋白质组学,已在Nature Commun、Cell Res、J am soc mass spectr、J proteome res等期刊发表学术论文12篇,综述2篇,获得发明专利一项

郑庆飞(通讯作者)
● 现任美国俄亥俄州立大学医学院放射肿瘤学系和癌症代谢中心助理教授,生物化学与药理学系兼职助理教授,博导
● 2012年本科毕业于清华大学化学系化学生物学基础科学班,2017年博士毕业于中国科学院上海有机化学研究所,2017-2021于美国纪念斯隆凯特琳癌症中心进行博士后研究工作,2021年8月加入俄亥俄州立大学。郑博士长期从事肿瘤微生物组、微生物代谢和癌症表观遗传学的研究,研究手段包括化学生物学、合成生物学、生物物理、化学蛋白质组学和代谢组学等。已发表科研论文50余篇,引用900余次,H-index 19,专利2项,担任Frontiers in Chemistry 和Frontiers in Molecular Biosciences化学生物学方向副主编,Molecules和Frontiers in Cellular and Infection Microbiology客座副主编,Biochemistry and Molecular Biology编委,iMeta和VIEW青年编委,曾获得第九届吴瑞奖等多个奖项。
更多推荐
(▼ 点击跳转)
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法
iMeta | 浙大倪艳组MetOrigin实现代谢物溯源和肠道微生物组与代谢组整合分析

第1卷第1期

第1卷第2期

第1卷第3期

第1卷第4期
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:office@imeta.science
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|




























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集




















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









