









 本文综述了GWAS(全基因组关联研究)后如何通过精细映射和基因优先排序来识别因果变异。GWAS标记变异通常位于高连锁不平衡区域,其中非编码变异可能在细胞类型和疾病特异性背景下影响基因表达。精细映射方法包括寻找功能元件重叠、等位基因特异性效应、转录因子结合位点破坏等。基因优先排序则通过eQTL、pQTL、mQTL等QTL分析来确定受影响的基因。尽管面临挑战,如一对一或多对多的因果关系复杂性,但这些方法为理解复杂性状的遗传基础提供了宝贵见解。
本文综述了GWAS(全基因组关联研究)后如何通过精细映射和基因优先排序来识别因果变异。GWAS标记变异通常位于高连锁不平衡区域,其中非编码变异可能在细胞类型和疾病特异性背景下影响基因表达。精细映射方法包括寻找功能元件重叠、等位基因特异性效应、转录因子结合位点破坏等。基因优先排序则通过eQTL、pQTL、mQTL等QTL分析来确定受影响的基因。尽管面临挑战,如一对一或多对多的因果关系复杂性,但这些方法为理解复杂性状的遗传基础提供了宝贵见解。
有了GWAS公共数据后的下一步就是找因果变异(causal varision),这篇文章介绍fine-mapping精细映射 和gene prioritization基因排序,简单一句话就是 translate GWAS loci to a functional understanding of the associated trait, while taking cell-type- and disease-specific context into account. 再简单点,搞清楚loci——trait的关系是不是因果。内容来自2020年1月Open Biology上发表的综述A practical view of fine-mapping and gene prioritization in the post-genome-wide association era。(注:涉及到因果就很麻烦,一对一,一对多,多对一,多对多,中介、调和、介导、反向等等等等,之后和孟德尔随机化、结构方程模型一起慢慢总结吧)。
表型性状phenotypic traits 和疾病都含有有影响其发育,易感性或特征的遗传成分。genetic regions (loci)基因座与表型性状相连接,GWAS通过比较和关联健康(baseline)人群和具有感兴趣性状特征(如1型糖尿病、乳糜泻和身高)的人群,获得数百万个相对常见的遗传变异(SNP)。通过GWAS获得的性状相关的遗传基因座,会被变异标记为一个marker或者top 变异。每个标记变异表示一个单体型,其中包含许多附近位于高度连锁不平衡(LD)变异,表明它们最有可能一起遗传。
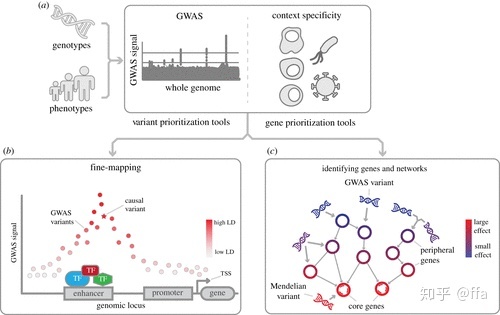
b:因果变异(星号)不是最强的GWAS信号,而是强LD中的变异,其顶部效应位于活性增强子区域。c:为了深入了解导致表型的生物学过程,可以对基因进行优先排序并建立因果网络。GWAS变异通常在人群中普遍存在,并且效应大小较小(蓝色)。它们影响的基因也更可能对表型产生很小的影响
下面两个原因导致因果变异很难被识别
(1)GWAS无法将标记变异信号与处于高LD的其他变异信号区分开。高LD(R2 > 0.8)中超过95%的变异位于非编码区,并且相距最多500 kb。它们中的任何一个都可能是实际的因果变量。
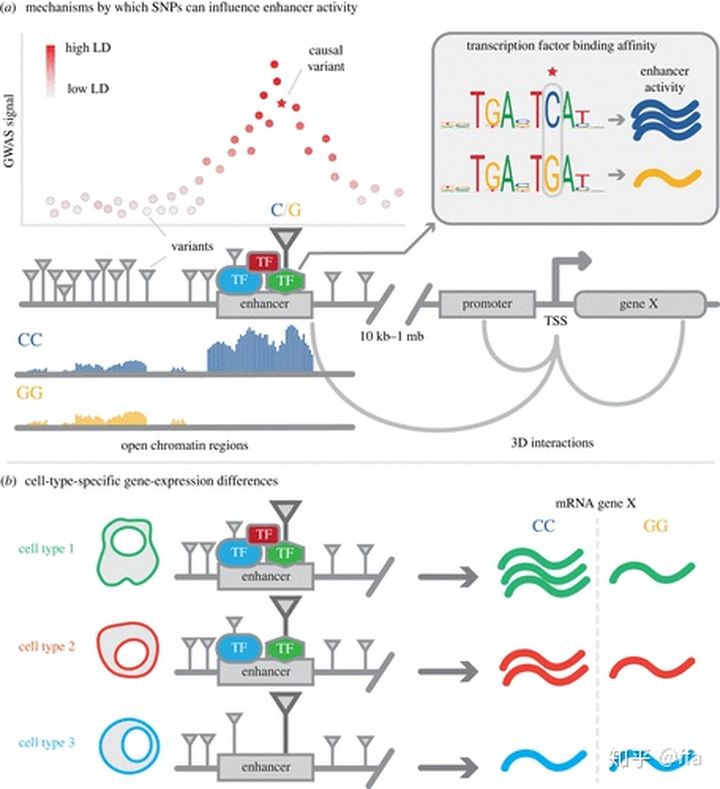
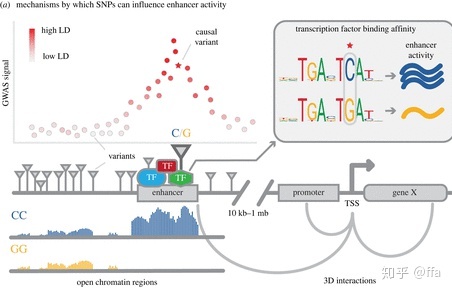
(2)非编码因果变异的影响可能是细胞类型,背景和疾病高度特异性的。细胞类型和组织以及疾病特定的微环境这三种因素会极大地影响涉及的变异、转录因子、基因和通路。非编码DNA包含可与转录因子(TF)蛋白结合并调节基因表达的调节区-增强子和启动子。使用哪种增强子和启动子取决于大约1600个人类TF的细胞类型特异性丰度及其在表观遗传学上对给定调控区域的可及性。遗传变异可以破坏任何这些TF的结合,从而导致增强子或启动子活性改变。反过来,这会影响基因表达和细胞通路。
大多数fine mapping方法都假设每个基因座仅单个变异会影响性状,其实这不符合实际情况,因为单个GWAS基因座中的多个变异也可能会影响单个基因的表达。有以下两种情况:(1)任一变异的作用以线性方式(加性效应)(2)两个或更多的需要来影响基因表达(上位性效应)。多种变异可能在单个基因座中发挥作用,multiple variants may play a role in a single locus这进一步使执行和解释精细映射和基因优先排序方法变得复杂。(Large-scale identification of sequence variants influencing human transcription factor occupancy in vivo. Nat. Genet. 47, 1393-1401. //Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell 167, 1369-1384.e19.) 涉及multiple variant, multiple triat的内容以后会经常碰到,其实假设过于简单的工具现在仍然在用。
Fine-mapping变异的方法共有五种:(1)找与功能元件的overlap;(2)利用等位基因特异性变异的效应;(3)找破坏TFBS转录因子结合位点的变异;(4)是直接测量变异对调控区域强度的影响;(5)利用3d交互找因果变异对基因表达的影响。
1.1 识别与功能元件重叠的变异
最直接的fine mapping是高LD 区域的GWAS变异与功能原件重叠(promoters,enhancers)的重叠。 The NIH Roadmap Epigenomics Mapping Consortium数据集有127个不同细胞系和组织的 ChIP-seq组蛋白修饰数据。Genetic and Epigenetic Fine-Mapping of Causal Autoimmune Disease Variants这篇论文对来自21种自身免疫性疾病的GWAS变异进行fime mapping,估计约有60%的候选因果变异映射到免疫细胞promoter,而另外约8%则映射到enhancer。这个研究的算法Probabilistic Identification of Causal SNPs (PICS)也比较有启发。文章2020年2个月也引了30次,要仔细读下。候选因果变体在CD4 + T细胞,CD8 + T细胞和B细胞的特定子集中富集再由组蛋白标记H3K27ac定义的增强子上。在单核细胞,中性粒细胞和CD4 + T细胞的另一项研究中也是如此。列一下另外几篇文献:Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat. Genet.2015Genetic drivers of epigenetic and transcriptional variation in human immune cells. Cell 2016Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat. Genet.2013
1.2 推断等位基因特异性变异的效应
基于等位基因分离包含变异的测序读段。测序读段的等位基因特异性丰度可以直接告知我们该变异在开放染色质区域上的功能。变异在调控区即causal等位基因失衡被称为染色质可及性的数量性状基因座(caQTL)。CD4 + T细胞ATAC-seq的peak中鉴定出许多caQTL,其在候选的causal自身免疫变异中富集。 通过改变与enhancer相关的H3K27ac或H3K4me1组蛋白修饰的peak影响调控区的变异或组蛋白QTL的存在,也暗示这些变异对细胞类型特异性enhancer的活性有影响。由于它们对DNA可及性和表观遗传标记的功能性影响,这些变异更可能是GWAS性状的因果变异。Fine-mapping cellular QTLs with RASQUAL and ATAC-seq. Nat. Genet. 48, 206-213.///Individuality and variation of personal regulomes in primary human T cells. Cell Syst. 1, 51-61.这两篇文献挺重要的。
非编码GWAS变异可以对基因表达产生等位基因作用的另一种机制是基因的可变剪接。GWAS相关变体具有诱导细胞类型特异性可变剪接(sQTL)的电势,或可能影响反式作用剪接调节基因(The expanding landscape of alternative splicing variation in human populations. Am. J. Hum. Genet. 102, 11-26.)。这在全基因组方法中得到了证实,该方法确定了622个具有内含子sQTL的外显子。这些外显子中有110个在LD中带有GWAS标记变异(Alternative splicing modulated by genetic variants demonstrates accelerated evolution regulated by highly conserved proteins. Genome Res. 26, 440-450.)。与多发性硬化症相关的PRKCA基因似乎受到内含子sQTL的影响,内含子sQTL增加了gene isoform的表达更倾向于无义介导的衰变,从而降低了转录后可能的保护性PRKCA mRNA水平。然而,sQTLs也似乎通过更复杂的机制起作用,例如通过caQTLs间接作用(RNA splicing is a primary link between genetic variation and disease. Science 352, 600-604.),或通过诱导替代性上游转录起始位点(Exon-mediated activation of transcription starts. bioRxiv 565184. )。这些表明sQTL可能是重要且复杂的机制,GWAS相关变体通过该机制影响性状。(这一部分研究相对于eQTL少得多,不太被人关注)查了下,有两个数据库可能有用:eQTL Catalogue(剪接数据比较专用),QTLbase(各种qtl)
1.3 关于tfbs的部分略
1.4 调控区域活性检测(看不懂没意思)
CAGE能够确定TSS的确切位置以及基因的表达水平,可以鉴定promoter和enhancer的效应,发现52% 的 promoter区变异效应再secondary CAGE peaks,说明“genes can have multiple active promoters depending on the genotype”(The effect of genetic variation on promoter usage and enhancer activity. Nat. Commun. 8, 1-7. )CAGE QTLs已被发现与系统性红斑狼疮(SLE)和炎性肠病有关的基因座,支持它们与免疫疾病的相关性。其余的测序方法(Reporter-plasmid assays.MPRA,STARR-seq ,SuRE)略
1.5 使用3D交互组从因果变体到基因
当鉴定出一个因果变异,变异对基因表达的影响可直接通过与它的靶基因调控区的影响和物理交互找到。这篇综述只讲了Hi-chip和Hi-c,其实最近还有Plac-seq这类技术。
使用HiChIP将包含自身免疫疾病优先变异的H3K27ac区与基因的TSS连接,并显示在Th17 中包含IL2基因的TSS 和rs7664452 之间的细胞类型特异性相互作用。记忆T细胞中的细胞和rs2300604与靶基因BATF之间。对于用HiChIP评估的684种自身免疫病相关变体,鉴定出2597个基因-变体相互作用,表明自身免疫病变体可以调控多种基因。此外,这些基因变异的相互作用中只有14%与最接近变异的基因有关。
因果变异的远程相互作用的另一个例子是rs1421085的相互作用,它与肥胖风险相关,位于FTO内含子中。TFBS破坏分析表明,rs1421085破坏了ARID5B TF结合基序并影响了调控IRX3和IRX5的增强子的活性,该基因位于上游1.2 Mb,而不是最初预期的共定位FTO基因本身。因此,精细映射和相互作用分析已经在与肥胖相关的风险基因座中发现了其他因果基因。
Hi-C识别特定启动子和增强基因相互作用。如通过各种原发性免疫细胞与类风湿性关节炎GWAS变异体的启动子-基因相互作用来优先处理四个类风湿性关节炎基因[ 19 ]。另一项研究分析了14种主要人体组织发现频繁相互作用的区域(FIREs)富集了疾病相关的GWAS变异。但由于分辨率限制,很难在调控区域内精确查明因果变异。此外,细胞类型和环境效应影响调控区域与基因的相互作用,仅在一种组织或细胞类型中发现了38.8%的FIRE 。因此,应结合公共数据库如EnhancerAtlas2.0 来找因果变异。
XGenetic variants regulating immune cell levels in health and disease
2利用GWAS特性对基因进行优先排序
fine mapping侧重于确定影响感兴趣性状的因果变异,但了解哪些变异是因果性的并不能确定该变异对性状的下游影响。通过识别受每个GWAS基因座影响的基因可以解决这个问题。此外,如果已知受基因座影响的因果基因,可以缩小潜在因果变异的范围。下图概括了4种排序的方法。
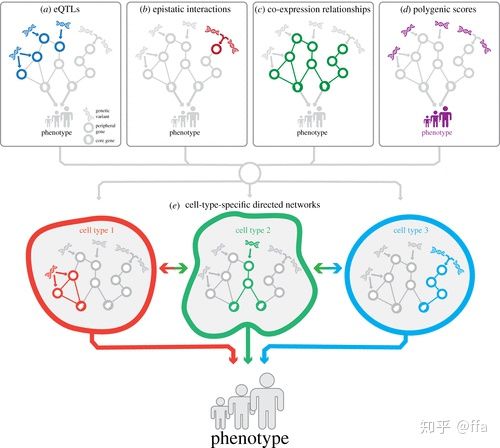
2.1 eQTL
caQTLs往往指示一个因果变异或调控区域,eQTL的特定子集可用于确定受一个GWAS基因座影响的基因。使用eQTL分析进行基因优先排序的最简单方法是overlap the marker variant of a GWAS locus with the top eQTL variant
更准确估计GWAS位点和QTL位点之间的重叠或因果关系的软件:包括FUMA,COLOC 和Mendelian Randomization。孟德尔随机化有很多改进方法,最常用的是SMR和MR-base两个,其他的都在bioRxiv上目前不知道好坏。(A novel Mendelian randomization method identifies causal relationships between gene expression and low-density lipoprotein cholesterol levels)。MR通常用于估计GWAS和QTL谱之间的因果关系并已成功鉴定与复杂性状有因果关系的基因。如SORT1对胆固醇水平、MR收到“multiple variants in LD can affect the same gene (linkage), and several genes can be affected by the same causal variants (pleiotropy)”的影响。 GCTA's COJO可以用来进行Independent variant selection
在mRNA表达的遗传力约70%归因于trans-eQTLs (An expanded view of complex traits: from polygenic to omnigenic. Cell 169, 1177-1186.//Trans effects on gene expression can drive omnigenic inheritance. Cell 177, 1022-1034.e6.)就是后面全基因模型那两篇论文。 trans-eQTL mapping做起来比较麻烦,所以平常大部分论文都只看到cis。用事先定义好的子集来做相对容易一些,见Distinctive roles of age, sex, and genetics in shaping transcriptional variation of human immune responses to microbial challenges. Proc. Natl Acad. Sci. USA 115, E488-E497. 这会错失一些基因。
不同的组织,细胞类型,时间点和刺激条件可能会诱导GWAS基因座中的许多不同表达模式以及与变异的不同相互作用。研究存在于除血液以外的组织的性状时,这是特别具有挑战性的,比如神经研究2019A gene co-expression network-based analysis of multiple brain tissues reveals novel genes and molecular pathways underlying major depression. PLoS Genet. 15, e1008245.,因为 cell-type- or context-specific QTL 研究不太多。最近有些进展: Unraveling the polygenic architecture of complex traits using blood eQTL metaanalysis. bioRxiv///Single-cell RNA sequencing identifies celltype-specific cis-eQTLs and co-expression QTLs. Nat. Genet. 50, 493-497. ///Genetic mapping of cell type specificity for complex traits. Nat. Commun. 10, 1-13.细胞分辨率的研究,TSPAN13和ZNF414上的eQTL所示,仅存在于CD4 + T细胞中,而不存在于bulk rna或其他细胞类型中。 Single-cell eQTLgen 和 the LifeTime consortium(这个计划感觉要凉)有这方面的数据。Single-cell eQTLGen Consortium: a personalized understanding of disease. arXiv
2.2 使用其他QTL识别GWAS基因座的下游影响
QTL有以下几种,上一篇讲了这种可以叫做xQTL,最近发现也有一些论文叫molQTL。
proteins (pQTL):Inter-individual variability and genetic influences on cytokine responses to bacteria and fungi. Nat. Med.2017. 22, 952-960. ///A functional genomics approach to understand variation in cytokine production in humans. Cell 167,2016, 1099-1110.e14. 对刺激的离体细胞因子反应已显示具有强大的遗传调控因子.发现的所有相关影响都是反(即不靠近细胞因子基因),这表明细胞因子的释放受受体途径中基因的控制,而不是直接受细胞因子mRNA水平的控制。此外,上下文特异性很重要,因为影响T细胞细胞因子的QTL被发现富含自身免疫GWAS基因座,而影响单核细胞细胞因子的QTL被感染疾病相关基因座更多。
metabolites (mQTL): Genome-wide study for circulating metabolites identifies 62 loci and reveals novel systemic effects of LPA. Nat. Commun.2016. 7, 1-9
DNA methylation (meQTL): Disease variants alter transcription factor levels and methylation of their binding sites. Nat. Genet.2017, 49, 131-138. 这里容易混淆,有的论文就叫mQTL。现在m开头的多了所以要细分。
microbiota (miQTL):Meta-analysis of human genome-microbiome association studies: the MiBioGen consortium initiative. Microbiome 6, 1-7.
cells (cell-count or ccQTL): XGenetic variants regulating immune cell levels in health and disease. Cell 2013, 242-256. ///Differential effects of environmental and genetic factors on T and B cell immune traits. Cell Rep. 2016
2.3 Crispr sceening的内容暂时不想看,略。
2.4 Mapping gene–gene regulatory interactions using population data(不好翻译直接英文)
共表达也可基于个体间的表达差异,其可用于疾病的基因的重要性排序,也可以找关于疾病的下游结果。DEPICT将基因共调控与GWAS数据整合在一起,以提供可能与该性状相关的因果基因和通路。GADO正确识别了83名孟德尔疾病患者的队列中41%的因果基因,并通过将特异性基因组与共表达相结合,对几个新的因果候选基因进行了优先排序网络。eMAGMA在大脑区域使用共表达和组织特异性eQTL来确定主要的抑郁症候群的99个致病基因。这些共表达模块富集于大脑区域,而不是全血,突出了共表达网络的组织特异性。
基于人群的共表达网络通过遗传学和环境描述了基因之间的关系。因此,仅基于共表达不可能将共表达的哪一部分归因于遗传。因此,这些网络用于精细映射因果变体的用途有限,主要用于在确定基因优先级后识别受GWAS基因座影响的基因和通路。
A gene co-expression network-based analysis of multiple brain tissues reveals novel genes and molecular pathways underlying major depression. PLoS Genet. 2019,15, e1008245.
An integrative approach for building personalized gene regulatory networks for precision medicine. Genome Med. 2018,10, 1-15.
Improving the diagnostic yield of exome-sequencing by predicting gene–phenotype associations using large-scale gene expression analysis. Nat. Commun.2019, 10, 1-13.
2.5 omnigenic model(之后会详细总结)
这个 omnigenic模型是Boyle EA在2017年Cell上提出的(An expanded view of complex traits: from polygenic to omnigenic.)。结果呢,澳洲那边的Jian Yang那一波人马上diss前面这个Core gene的概念。但截止2020年2月Boyle的第一篇文章已经引了900快1000次了,可见还是有一定接受度的。19年5月第一篇论文的同一拨人Pritchard又发了篇Cell改进模型“Trans Effects on Gene Expression Can Drive Omnigenic Inheritance”,目前还没看到有后续跟进。
以身高为例,受多基因影响,大多数100 kb基因组窗口似乎有助于解释其变异。鉴于个体变异的影响大小也越来越如此之小,它提出了一个问题:什么是个体变异平均的复杂性状的因果关系?如果Omnigenic模型是正确的,将对精细映射GWAS基因座提出重大挑战,特别是对于下游变异结果的解释。因为遗传对性状的影响只会增加。除此之外,当前的功能测定可能不适合模拟在数百万个人群的人口研究中观察到的微小和微妙的变异效应以及基因-基因或基因-环境相互作用。
来自与性状相关的所有基因座的完整GWAS信号可用于估计多基因分数(PGS),该分数描述个体对给定性状的遗传易感性。在其最基本的形式中,PGS构成了所有独立风险基因型的线性组合,并由GWAS效应大小加权。对于性状的PGS可以与基因(和蛋白质)的群体中的表达水平相关联。如果存在强相关性,则以PGS为代表的GWAS基因座共同影响这些基因。这些基因可能代表与疾病相关的共表达网络中的核心基因。Unraveling the polygenic architecture of complex traits using blood eQTL metaanalysis这篇文章挂在bioRxiv上一年多迟迟没有发表,但里面的结果很好地诠释了上面这段话,需要重视。里面也提到了PGS and gene expression (expression quantitative trait score, eQTS)这个概念,把多基因评分和QTL也联系起来。另外一篇是“Integration of multi-omics data and deep phenotyping enables prediction of cytokine responses.”也是这类。还有篇文章“Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018,50, 1219-1224. ”也很重要。
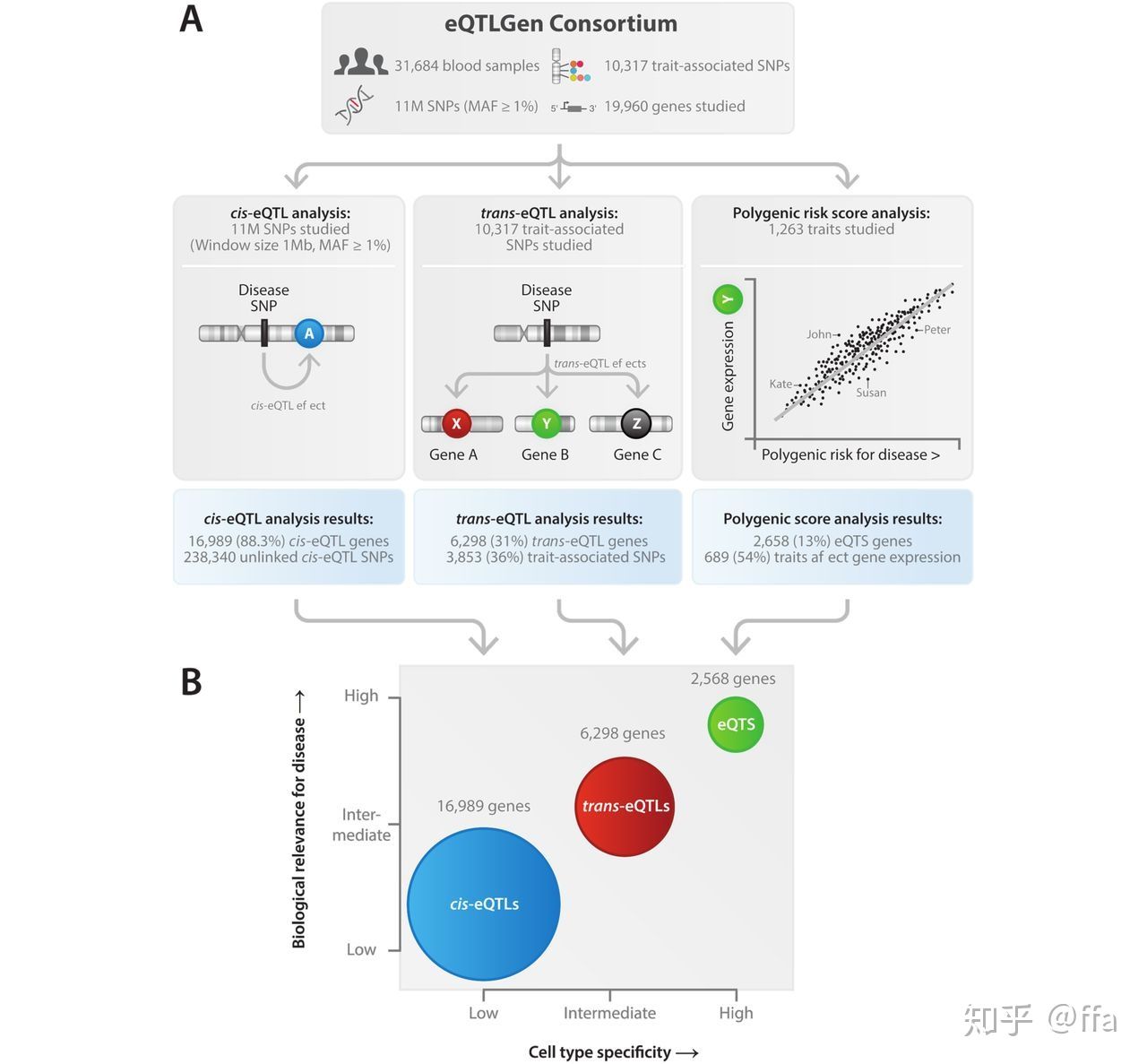
鉴于我们已经意识到性状的可能是多基因的,甚至是全基因的,因此精细映射单个GWAS基因座似乎是不可能的任务。也许可能无法完全理解一个性状的所有微小影响和相互作用,但通过使用抽象层如PGS这类指标,可以推断出这些效应的共同结果。也就是说,PGS不单单可以用于临床预测,也可以在生命科学研究中用于推断变异对性状的共同影响(这里也能想到,类似找一些metagene或者各种评分的方法,可能也是有效的。不确定这个想法对不对)。the genes and pathways associated with stronger or joint genetic effects are more likely candidates for drug interventions. 这句话看评审意见是审稿人硬让加上去的,姑且看一下。
最后要说注意事项了:(1)这些方法单一使用不足以识别候选变体和基因的全部因果关系。看到哪篇文章写过,最好综合好几种方法的结果一起来看,但看发表的论文好像没有这么做的。(2)目前数据集中在血液组织,已经识别出许多与性状相关的潜在因果变体和基因。但需要使用组织和细胞类型特有的资源以及特异于性状的环境因素来完善和验证这些候选物,尽可能地概括每个特征的真实生物学状态。(3)优先基因可能不存在,也不是实际的药物靶标。(4)结果可用于生成预测模型,快速,非侵入性地识别普通人群中特异性变异和基因。(5)找到调控区域及其下游基因。
最后的最后,这篇文章对triat和loci和variant的数量没有做太多说明,其实是挺关键的一个问题,后面再讲。
发布于 2020-02-23

















 2万+
2万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









