







 本文探讨了第一性原理计算在电池材料研究中的应用,特别是通过VASP软件计算氧空位和结合能。文章详细介绍了计算参数的选取,如截断能、k点和泛函,并阐述了氧空位在电极材料稳定性中的角色。研究表明,氧空位形成能的高低影响电池性能,而结合能则反映材料的结构稳定性。
本文探讨了第一性原理计算在电池材料研究中的应用,特别是通过VASP软件计算氧空位和结合能。文章详细介绍了计算参数的选取,如截断能、k点和泛函,并阐述了氧空位在电极材料稳定性中的角色。研究表明,氧空位形成能的高低影响电池性能,而结合能则反映材料的结构稳定性。
概述
广义的第一原理包括两大类,以 Hartree-Fork 自洽场计算为基础的 ab initio 从头算和密度泛函理论(DFT)计算。也有人主张,ab initio 专指从头算,而第一性原理和所谓量子化学计算特指密度泛函理论计算。
第一性原理是指将多原子构成的体系理解为由电子和原子核组成的多粒子系统,在解体系薛定谔方程的过程中,最大限度地进行“非经验性”处理,即不涉及任何经验参数,所要输入的只是原子的核电荷数和一些模拟环境参量。
计算所求得的结果是体系薛定谔方程的本征值和本征函数 (波函数),有了这两项结果,就可研究体系的基本物理性质。
密度泛函理论(DFT)在许许多多的研究人员不断努力中得到了质的飞跃。随着现代科技的进步,尤其是芯片技术不断更新迭代使得计算机性能也得到了质的飞跃,并且人们对物理理论的认识也更加的深入。计算机模拟对材料进行设计是当代科学研究不可或缺的一种研究手段。相比较传统的实验方法,在许多情况下计算机模拟比实验更快、更省,并且还可以预测一些当前实验水平难以达到的理想情况。密度泛函的计算迭代过程如图一所示。
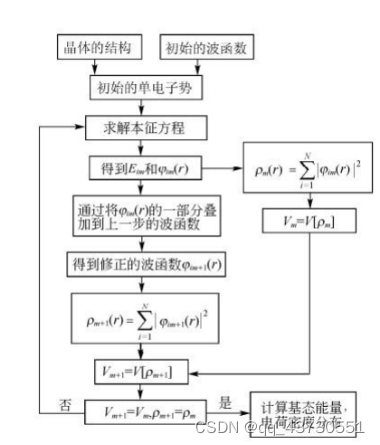
图1 密度泛函理论计算迭代过程示意图
密度泛函在电池基础研究中有着广泛的应用,常用于计算电极材料的结构稳定性、嵌锂电位、电子结构、能带、弛豫结构、缺陷生成能、迁移路径、活化能以及锂离子传输动力学和脱嵌锂相变等性质。本文主要讲述使用VASP的计算流程,并简要分析结合能和氧空位。
计算重要参数
在理论计算过程中,输入文件是必不可少的,其参数的设定也尤为重要。
2.1 计算过程中的主要参数
计算主要采用的是 VASP.5.4.4 程序,采用面波基组对SFT、SFM进行计算,为得到准确结果的同时又节约时间,需要对相关参数进行测试,需要测试的参数主要包括截断能、k 点以及泛函。赝势选择 VASP 自带的 PAW 势,每个原子的能量收敛标准为 0.0001 eV,每个原子的自洽收敛标准为 10-5 eV。由于体系中 Fe 元素为磁性元素,计算时应将自旋极化考虑在内。
使用VASP计算需要四个输入文件,INCAR文件控制了VASP进行何种性质的计算;POSCAR文件描述了所计算的体系的晶胞参数(包括基矢、晶格常数、原子类型、坐标位置);POTCAR文件包含了体系中
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 5581
5581

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









