






 本文介绍了使用R语言进行单细胞RNA测序数据分析的过程,包括数据读取、矩阵转换、Seurat对象创建、数据过滤、线粒体基因比例检查、测序深度相关性分析以及基因变异性筛选。通过小提琴图和散点图展示数据质量,并对高线粒体比例的细胞进行排除,确保后续分析的准确性。最后,进行了数据标准化和变量基因的识别。
本文介绍了使用R语言进行单细胞RNA测序数据分析的过程,包括数据读取、矩阵转换、Seurat对象创建、数据过滤、线粒体基因比例检查、测序深度相关性分析以及基因变异性筛选。通过小提琴图和散点图展示数据质量,并对高线粒体比例的细胞进行排除,确保后续分析的准确性。最后,进行了数据标准化和变量基因的识别。
质控和数据过滤
准备工具:R。
准备数据:上期经过整理的数据geneMatrix。
注意事项:R的安装目录和文件所在位置都不可有英文。
R 语言所需安装的包:
#if (!requireNamespace("BiocManager", quietly = TRUE))
# install.packages("BiocManager")
#BiocManager::install("singscore")
#BiocManager::install("GSVA")
#BiocManager::install("GSEABase")
#BiocManager::install("limma")
#BiocManager的包安装命令可能会改变,需要去BiocManager官网查询最新安装方法,短时间内不会改变的。
#install.packages("devtools")
#library(devtools)
#devtools::install_github('dviraran/SingleR')
#这个包作用为对细胞的注释,对富集分析作用很关键,在这一篇章可以先不安装,在以后的go富集分析才需要。
正文代码开始:`#读取数据
library(limma)
library(Seurat)
library(dplyr)
library(magrittr)
setwd("数据所在目录") #设置工作目录
#读取文件,并对重复基因取均值
rt=read.table("geneMatrix.txt",sep="\t",header=T,check.names=F)
rt=as.matrix(rt)
rownames(rt)=rt[,1]
exp=rt[,2:ncol(rt)]
dimnames=list(rownames(exp),colnames(exp))
data=matrix(as.numeric(as.matrix(exp)),nrow=nrow(exp),dimnames=dimnames)
data=avereps(data)#如果基因存在多行中,为了取得结果就会取均值
#将矩阵转换为Seurat对象,并对数据进行过滤
pbmc <- CreateSeuratObject(counts = data,project = "seurat", min.cells = 3, min.features = 50, names.delim = "_",)#min.cells为基因存在样本最小数,需要根据实际情况选择,min.features = 50基因最小存在细胞数
#使用PercentageFeatureSet函数计算线粒体基因的百分比
pbmc[["percent.mt"]] <- PercentageFeatureSet(object = pbmc, pattern = "^MT-")
pdf(file="04.featureViolin.pdf",width=10,height=6) #保存对于基因特征的小提琴图
VlnPlot(object = pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
dev.off()
pbmc <- subset(x = pbmc, subset = nFeature_RNA > 50 & percent.mt < 7) #对数据进行过滤,线粒体比例大于7%进行删掉,因为线粒体基因比例过高的细胞,会干扰细胞分群
#测序深度的相关性绘图
pdf(file="04.featureCor.pdf",width=10,height=6) #保存基因特征相关性图
plot1 <- FeatureScatter(object = pbmc, feature1 = "nCount_RNA", feature2 = "percent.mt",pt.size=1.5)
plot2 <- FeatureScatter(object = pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA",,pt.size=1.5)
CombinePlots(plots = list(plot1, plot2))
dev.off()
#对数据进行标准化
pbmc <- NormalizeData(object = pbmc, normalization.method = "LogNormalize", scale.factor = 10000)
#提取那些在细胞间变异系数较大的基因
pbmc <- FindVariableFeatures(object = pbmc, selection.method = "vst", nfeatures = 1500)
#输出特征方差图
top10 <- head(x = VariableFeatures(object = pbmc), 10)
pdf(file="04.featureVar.pdf",width=10,height=6) #保存基因特征方差图
plot1 <- VariableFeaturePlot(object = pbmc)
plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE)#top10是对基因
CombinePlots(plots = list(plot1, plot2))
dev.off()`
结果
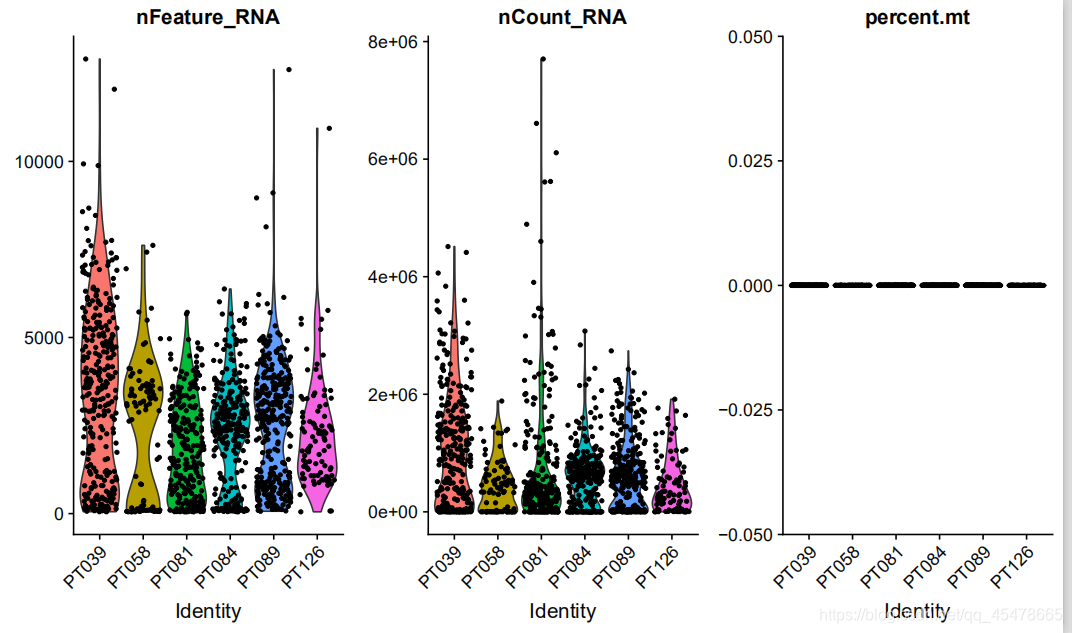
这是小提琴图,第一个图是在样品中对所有细胞检测到的基因数量小提琴图,横坐标为样品名,纵坐标为每个细胞中包含的基因数量;第二个图为样品所有细胞的中基因序列数量小提琴图,横坐标为样品名,纵坐标为每个细胞基因的数量。第三个图为样品所有细胞的线粒体比例小提琴图,横坐标为样品名,纵坐标为每个细胞线粒体比例。小提琴图展示了任意位置的密度,可以知道哪些位置的密度较高。
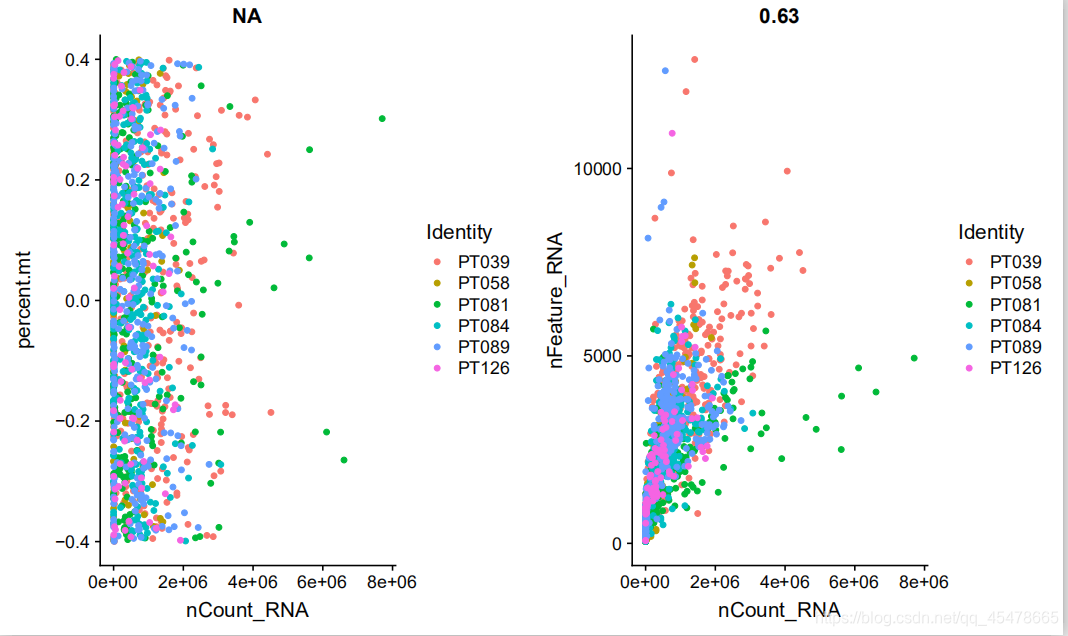
随着测序数量的增加,单细胞所检测到的基因数量也在增加,其中,这两个是有一定关联性的,在作图之前需要对含有线粒体基因的细胞进行筛选,因为过高会干扰细胞分群,我所选择的是高于7%的进行筛选,第一个图是对于线粒体比例于测序数据的关系的散点图即测序深度和线粒体基因含量的关系,第二个是对基因于测序数据序列的关系的散点图,即测序深度于基因数量的关系。
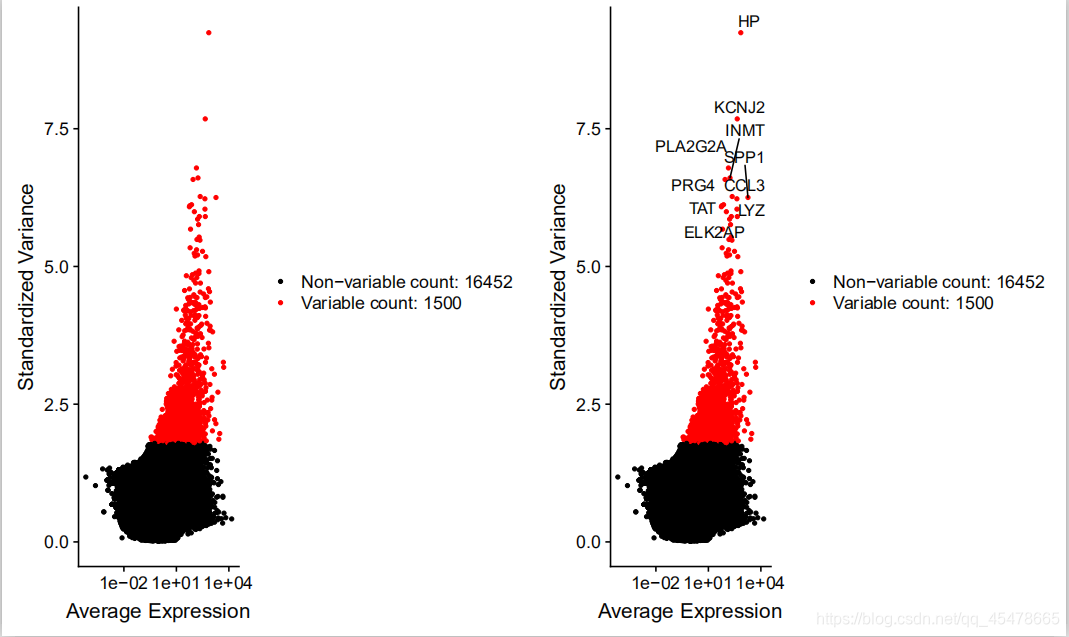
基因离差散点图,第一个图,是进行筛选,是筛选基因表达最高的前1500个(表达量要根据实际情况进行更改),在这个图中横坐标是对于基因在所有细胞的平均表达值,纵坐标是对于基因的离差值,离差值值越大表示基因的可变性越大。
第二个图是对表达最显著的前十个基因进行标注。
单细胞测序流程(三)质控和数据过滤到这里就结束了
下一章会讲解单细胞的PCA主成分分析
我所做的所有分析与教程的代码都会在我的个人公众号中,请打开微信搜索“生信学徒”进行关注,欢迎生信的研究人员和同学前来讨论分析。
ps:公众号刚刚建立比较简陋,但是该有的内容都不会少。

















 9838
9838

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









