






生信分析中富集分析是必不可少的,最常见是调用clusterProfiler包进行分析。clusterProfiler包功能也比较强大,主要是做GO和KEGG的功能富集及其可视化。
但clusterProfiler包依赖的包很多,不仅安装过程复杂、耗时长,而且经常出现各种各样的错误。对于生信小白来说,错误很难解决。本文总结常见的报错。
clusterProfiler包的安装:
R version 3.6以上版本:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("clusterProfiler")R version 3.6以下版本:
source("http://bioconductor.org/biocLite.R")
biocLite()
biocLite("clusterProfiler")按上述的方法安装很少出现问题了。
安装报错的情况和解决办法:
1、clusterProfiler包使用有很多依赖包,经常出现依赖包安装失败情况(如下图)。解决办法也很简单:直接下载安装缺少的依赖包,下图的情况直接安装ggridges包,再library就可以正常使用。

2、文件夹权限问题,导致部分文件不能读入。解决方法:需要重新设置R文件夹读入权限
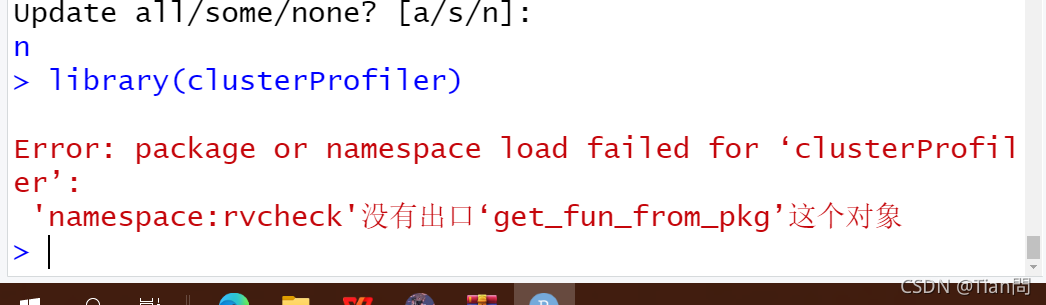
本人安装过程,出现这个报错,开始重新安装R和Rstudio也不行。最后升级为R-4.1.1版本终于在R中安装成功,但是在Rstudio中不能library(本人重新更改了包的安装路径)。最后在将下载完成的依赖包解压缩到Rstudio中发现部分文件不能读入,发现了权限问题。
! C:\Users\songyuyang\AppData\Local\Temp\RtmpGqkO4G\downloaded_packages\XVector_0.32.0.zip: 无法创建 F:\R-4.1.1\library\XVector\libs\x64\XVector.dll
拒绝访问。
! C:\Users\songyuyang\AppData\Local\Temp\RtmpGqkO4G\downloaded_packages\zlibbioc_1.38.0.zip: 无法创建 F:\R-4.1.1\library\zlibbioc\libs\x64\zlib1bioc.dll
拒绝访问。
! C:\Users\songyuyang\AppData\Local\Temp\RtmpGqkO4G\downloaded_packages\ape_5.5.zip: 无法创建 F:\R-4.1.1\library\ape\libs\x64\ape.dll
拒绝访问。
解决办法:
先进入自己的R安装位置,如我的为R-4.1.1,进入属性


进入安全,在点击编辑,将所有的组或用户名全改为允许,在应用。

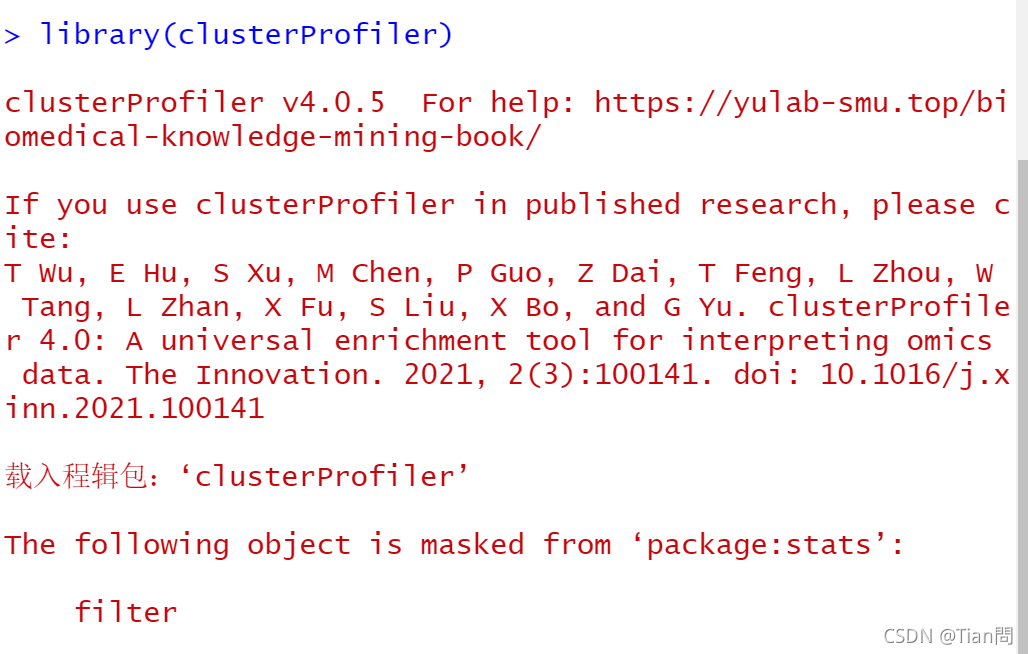
最后按照文章开始下载clusterProfiler并安装,安装成功如下图:














 713
713











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









