





 本文介绍了一种通过整合多个独立数据集来分析特定基因(如MMP7)与特发性肺纤维化(IPF)患者生存期关联的方法。通过对表达数据进行预处理和标准化,构建了用于生存分析的phedata数据集,并利用survival和survminer包进行了批量基因的存活曲线分析。
本文介绍了一种通过整合多个独立数据集来分析特定基因(如MMP7)与特发性肺纤维化(IPF)患者生存期关联的方法。通过对表达数据进行预处理和标准化,构建了用于生存分析的phedata数据集,并利用survival和survminer包进行了批量基因的存活曲线分析。
制作IPF基因集合 用于分析某个基因是否与生存期相关
load("G:/r/duqiang_IPF/surval_analysis_3_independent_dataset_IPF/combined_data_for_surval.RDdata")
1#输入想要查询的基因名称或者向量
gene_interested="MMP7" #输入想要查询的基因名称或者向量
library(stringr)
gene_interested=readClipboard() %>% str_split(pattern = ",",gene_interested)[[1]]
2#首先查看基因是否存在数据集中,如果不存在则去掉该基因
table( gene_interested %in% rownames(expr.17077clean) &
gene_interested %in% rownames(expr.freibrug.IPF))
#制作phedata数据用于存活分析
if(1==1){#制作phedata数据用于存活分析
for (eachgene in gene_interested) {
phe.freigbrug[paste0(eachgene)]=ifelse(expr.freibrug.IPF[eachgene,]>median(expr.freibrug.IPF[eachgene,]),
"High",'Low')
}
head(phe.freigbrug)
for (eachgene in gene_interested) {
phe.senia[paste0(eachgene)]=ifelse(expr.siena.IPF[eachgene,]>median(expr.siena.IPF[eachgene,]),
"High",'Low')
}
head(phe.senia)
for (eachgene in gene_interested) {
phe.17077[paste0(eachgene)]=ifelse(expr.17077clean[eachgene,]>median(expr.17077clean[eachgene,]),
"High",'Low')
}
head(phe.17077)
}
###开始合并三个数据集的phe数据
phe_final_3=rbind(phe.freigbrug,phe.senia,phe.17077)
dim(phe_final_3) #[1] 176 5
head(phe_final_3)
library(dplyr)
phe_final_3=phe_final_3 %>% transform(time=as.numeric(time))%>% transform(event=as.numeric(event))
getwd()
#批量基因差异分析
library(survival)
library(survminer)
for (eachgene in gene_interested) {
p=ggsurvplot(survfit(Surv(time, event)~phe_final_3[,eachgene],
data=phe_final_3), conf.int=F, pval=TRUE)
pdf(paste0(eachgene, "_surval_analysis_from_3_institutes.pdf"),width = 5, height = 5)
print(p, newpage = FALSE)
dev.off()
}
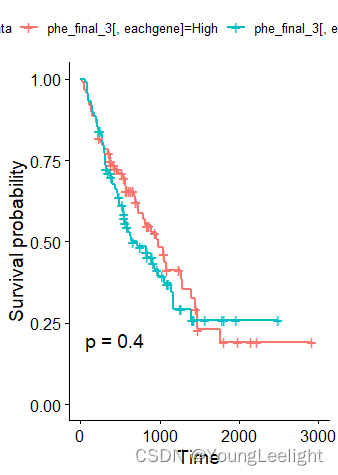
THBS2
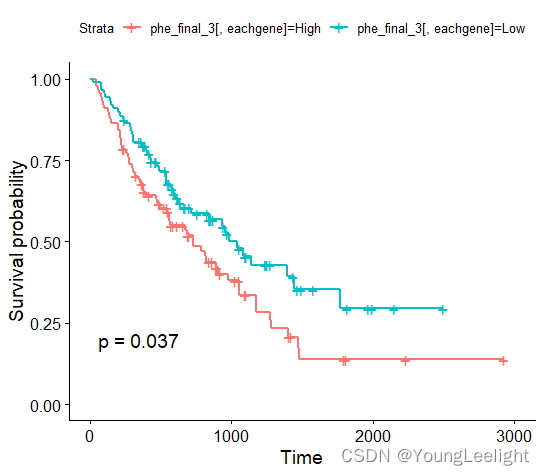
ASB2
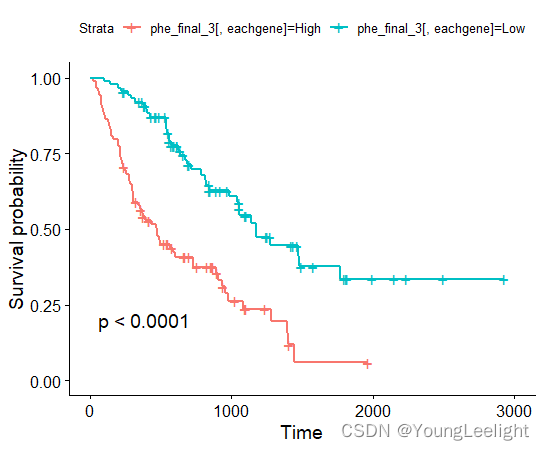
MMP7
if(1==1){
#读取感兴趣的基因
gene_interested=readClipboard()
head(gene_interested)
library(stringr)
gene_interested=str_split(pattern = ",",gene_interested)[[1]]
gene_interested=gene_interested[-which(gene_interested=="RAB40A")]
gene_interested
#gpl14550
load(file ="G:/r/duqiang_IPF/GSE70866—true—_BAL_IPF_donors_RNA-seq/Rdatafor_freibrug.RData")
head(expr.freiburg_clean)[,1:4]
head(meta.14550)[,1:4]
dim(expr.freiburg)
dim(meta.14550)
exprSet.114550.ipf=expr.freiburg[,which(colnames(expr.freiburg)=="GSM1820739"):which(colnames(expr.freiburg)=="GSM1820850")]
dim(exprSet.114550.ipf) #[1] 20330 112
head(exprSet.114550.ipf)[,1:4]
#ID 转换
if(1==1){
ids14550=data.table::fread("G:/r/duqiang_IPF/GSE70866_BAL_IPF_donors_RNA-seq/GPL14550-9757.txt",
)##读取
head(ids14550)
colnames(ids14550)
ids14550=ids14550[,c("ID","GENE","GENE_SYMBOL")]
head(ids14550)
colnames(ids14550) <- c("PROBE_ID","Entrez_ID", "SYMBOL_ID")#改名,让他适合下面的自定义函数
#自建函数
p2g <- function(eset,probe2symbol){
library(dplyr)
library(tibble)
library(tidyr)
eset <- as.data.frame(eset)
p2g_eset <- eset %>%
rownames_to_column(var="PROBE_ID") %>% #合并探针的信息
inner_join(probe2symbol,by="PROBE_ID") %>% #去掉多余信息
select(-PROBE_ID) %>% #重新排列
dplyr::select(SYMBOL_ID,everything()) %>% #求出平均数(这边的点号代表上一步产出的数据)
mutate(rowMean = rowMeans(.[grep("GSM", names(.))])) %>% #去除symbol中的NA
filter(SYMBOL_ID != "NA") %>% #把表达量的平均值按从大到小排序
arrange(desc(rowMean)) %>% # symbol留下第一个
distinct(SYMBOL_ID,.keep_all = T) %>% #反向选择去除rowMean这一列
dplyr::select(-rowMean) %>% # 列名变成行名
column_to_rownames(var = "SYMBOL_ID")
#save(p2g_eset, file = "p2g_eset.Rdata")
return(p2g_eset)
}
p2g_eset <- p2g(eset = exprSet.114550.ipf, probe2symbol = ids14550)
head(p2g_eset)
exprSet.114550.ipf=p2g_eset[,!colnames(p2g_eset)=="Entrez_ID"]
}
head(exprSet.114550.ipf)[,1:4]
colnames(meta.14550)=c('event','time','sex','diagnosis')
head(meta.14550)[,1:4]
meta.14550=meta.14550[rownames(meta.14550) %in% colnames(exprSet.114550.ipf),]
head(meta.14550)[,1:4]
dim(meta.14550) #[1] 112 7
dim(exprSet.114550.ipf) #[1] 20330 112
head(exprSet.114550.ipf)[,1:4]
phe.14550=transform(meta.14550,event=as.numeric(event)) %>% transform(time=as.numeric(time))
phe.14550=phe.14550[,1:3]
head(phe.14550)
exprSet.114550=exprSet.114550.ipf %>% transform(as.numeric()) %>% as.matrix()
head(exprSet.114550)[,1:3]
for (eachgene in gene_interested) {
phe.14550[paste0(eachgene)]=ifelse(exprSet.114550[eachgene,]>median(exprSet.114550[eachgene,]),
"High",'Low')
}
head(phe.14550)
dim(phe.14550)
dim(phe.17077)
##gpl17077
load(file ="G:/r/duqiang_IPF/GSE70866—true—_BAL_IPF_donors_RNA-seq/expr17077.RData")
head(expr.17077clean)
dim(expr.17077clean) #[1] 20190 64
head(meta.17077)
colnames(meta.17077)=colnames(meta.14550)
head(meta.17077)
meta.17077=meta.17077[,1:3]
head(meta.17077)
head(expr.17077clean)[,1:3]
library(dplyr)
phe.17077=meta.17077
head(meta.17077)
exprSet.17077=expr.17077clean %>% as.matrix() %>% transform(as.numeric()) #数据格式转换
head(exprSet.17077)[,1:3]
for (eachgene in gene_interested) {
phe.17077[paste0(eachgene)]=ifelse(exprSet.17077[eachgene,]>median(exprSet.17077[eachgene,]),
"High",'Low')
}
head(phe.17077)
##开始合并两个平台的phe数据
phe_final_3=rbind(phe.14550,phe.17077)
dim(phe_final_3) #[1] 176 37
getwd()
dir.create("G:/r/duqiang_IPF/GSE70866—true—_BAL_IPF_donors_RNA-seq/survival_for_genes-three")
setwd("G:/r/duqiang_IPF/GSE70866—true—_BAL_IPF_donors_RNA-seq/survival_for_genes-three")
head(phe_final_3)
phe_final_3=phe_final_3 %>% transform(time=as.numeric(time))%>% transform(event=as.numeric(event))
getwd()
#save(phe_final_3,meta.14550,meta.17077,expr.17077clean,exprSet.114550,file = "G:/r/duqiang_IPF/GSE70866—true—_BAL_IPF_donors_RNA-seq/survival_for_genes-three/3-institutes.RData")
load("G:/r/duqiang_IPF/GSE70866—true—_BAL_IPF_donors_RNA-seq/survival_for_genes-three/3-institutes.RData")
#批量基因差异分析
for (eachgene in gene_interested) {
p=ggsurvplot(survfit(Surv(time, event)~phe_final_3[,eachgene],
data=phe_final_3), conf.int=F, pval=TRUE)
pdf(paste0(eachgene, "_surval_analysis_from_3_institutes.pdf"),width = 5, height = 5)
print(p, newpage = FALSE)
dev.off()
}
load("G:/r/duqiang_IPF/GSE70866—true—_BAL_IPF_donors_RNA-seq/survival_for_genes-three/3-institutes.RData")
#批量基因差异分析
head(phe_final_3)
}
load("G:/r/duqiang_IPF/GSE70866—true—_BAL_IPF_donors_RNA-seq/survival_for_genes-three/3-institutes.RData")
colnames(exprSet.114550)
nrow(meta.14550)
dim(exprSet.114550) #[1] 20190 112
if(1==1){
head(meta.14550)
expr.freibrug.IPF=exprSet.114550[,which(colnames(exprSet.114550)=="GSM1820739"):which(colnames(exprSet.114550)=="GSM1820800")]
meta.freibrug.IPF=meta.14550[1:62,]
expr.siena.IPF=exprSet.114550[,!(colnames(exprSet.114550) %in% colnames(expr.freibrug.IPF)) ]
meta.siena.IPF=meta.14550[rownames(meta.14550) %in% colnames(expr.siena.IPF),]
head(meta.siena.IPF)
dim(meta.siena.IPF) #[1] 50 7
dim(expr.17077clean) #[1] 20190 64
head(meta.17077)
colnames(meta.17077)=c("time","event","sex","diagnosis")
head(meta.17077)
meta.17077=meta.17077[,1:4] %>%select(event,everything())
meta.14550=meta.14550[,1:4]
head(meta.14550)
meta.freibrug.IPF=meta.freibrug.IPF[,1:4]
meta.siena.IPF=meta.siena.IPF[,1:4]
head(meta.17077)
dim(expr.17077clean)# [1] 20190 64
identical(rownames(expr.freibrug.IPF),
rownames(expr.17077clean))
phe.freigbrug=meta.freibrug.IPF
phe.senia=meta.siena.IPF
phe.17077=meta.17077
expr.17077clean=as.matrix(expr.17077clean)
getwd()
dir.create("G:/r/duqiang_IPF/surval_analysis_3_independent_dataset_IPF")
setwd("G:/r/duqiang_IPF/surval_analysis_3_independent_dataset_IPF")
save(expr.freibrug.IPF, phe.freigbrug,
expr.siena.IPF, phe.senia,
expr.17077clean, phe.17077,
file ="G:/r/duqiang_IPF/surval_analysis_3_independent_dataset_IPF/combined_data_for_surval.RDdata" )
}




















 1551
1551

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











