






简介
在单细胞基因组学领域,将新数据集映射到已建立的参考数据集上的能力,与读取映射工具变革基因组序列分析的方式如出一辙。
一个关键的挑战是将这种参考映射框架扩展到不测量基因表达的技术,即使底层参考是基于单细胞RNA测序(scRNA-seq)的。在Hao et al, Nat Biotechnol 2023中,介绍了“桥接整合”(bridge integration),它使得将补充技术(如单细胞ATAC-seq(scATAC-seq)、单细胞DNA甲基化(scDNAme)、细胞因子分析(CyTOF))映射到scRNA-seq参考数据上成为可能,使用一个“多组学”数据集作为分子桥接。
在本文中,我们将展示如何将人类PBMC的scATAC-seq数据集映射到我们之前构建的PBMC参考数据。我们使用一个公开可用的10x多组学数据集作为桥接数据集,该数据集在同一细胞中同时测量基因表达和染色质可及性。
我们展示了以下内容:
-
加载和预处理scATAC-seq、多组学和scRNA-seq参考数据集 -
通过桥接整合映射scATAC-seq数据集 -
探索和评估所得注释
Azimuth ATAC用于桥接整合
用户现在可以在Azimuth网站或R中使用新发布的Azimuth ATAC工作流程自动运行PBMC和骨髓scATAC-seq查询的桥接整合。有关在R中本地运行的更多详细信息,请参见此vignette中的ATAC数据部分。
library(Seurat)
library(Signac)
library(EnsDb.Hsapiens.v86)
library(dplyr)
library(ggplot2)
加载桥接、查询和参考数据集
我们首先加载一个包含约12,000个健康捐献者的PBMC的10x多组学数据集。该数据集在同一细胞中测量RNA-seq和ATAC-seq,并可从10x Genomics 此处下载。我们遵循Signac包vignette中的加载说明。请注意,使用Signac时,请确保您使用的是最新版本的Bioconductor,因为用户在使用较旧的BioC版本时报告了错误。
加载和设置10x多组学对象
# 10x hdf5文件包含两种数据类型。
inputdata.10x <- Read10X_h5("/brahms/hartmana/vignette_data/pbmc_cellranger_arc_2/pbmc_granulocyte_sorted_10k_filtered_feature_bc_matrix.h5")
# 提取RNA和ATAC数据
rna_counts <- inputdata.10x$`Gene Expression`
atac_counts <- inputdata.10x$Peaks
# 创建Seurat对象
obj.multi <- CreateSeuratObject(counts = rna_counts)
# 获取线粒体基因的百分比
obj.multi[["percent.mt"]] <- PercentageFeatureSet(obj.multi, pattern = "^MT-")
# 添加ATAC-seq检测
grange.counts <- StringToGRanges(rownames(atac_counts), sep = c(":", "-"))
grange.use <- seqnames(grange.counts) %in% standardChromosomes(grange.counts)
atac_counts <- atac_counts[as.vector(grange.use), ]
# 获取基因注释
annotations <- GetGRangesFromEnsDb(ensdb = EnsDb.Hsapiens.v86)
# 切换到UCSC风格
seqlevelsStyle(annotations) <- 'UCSC'
genome(annotations) <- "hg38"
# 包含ATAC每个片段信息的文件
frag.file <- "/brahms/hartmana/vignette_data/pbmc_cellranger_arc_2/pbmc_granulocyte_sorted_10k_atac_fragments.tsv.gz"
# 将ATAC-seq数据作为ChromatinAssay对象添加
chrom_assay <- CreateChromatinAssay(
counts = atac_counts,
sep = c(":", "-"),
genome = 'hg38',
fragments = frag.file,
min.cells = 10,
annotation = annotations)
# 将ATAC检测添加到多组学对象
obj.multi[["ATAC"]] <- chrom_assay
# 根据QC指标过滤ATAC数据
obj.multi <- subset(
x = obj.multi,
subset = nCount_ATAC < 7e4 &
nCount_ATAC > 5e3 &
nCount_RNA < 25000 &
nCount_RNA > 1000 &
percent.mt < 20)
scATAC-seq 查询数据集代表来自健康捐赠者的约 10,000 个 PBMC。我们加载峰/细胞矩阵,存储片段文件的路径,并向对象添加基因注释,遵循多组实验中 ATAC 数据的步骤。
加载并设置 10x scATAC-seq 查询
我们从最近的论文中加载 reference:
obj.rna <- readRDS("/brahms/haoy/seurat4_pbmc/pbmc_multimodal_2023.rds")
映射scATAC-seq数据集使用桥接整合
现在我们已经设置了参考、查询和桥接数据集,我们可以开始整合。桥接数据集使得scRNA-seq参考和scATAC-seq查询之间的转换成为可能,有效地扩展了参考数据集,使其能够映射新的数据类型。
我们称这个扩展的参考为“扩展参考”,首先设置它。请注意,您可以保存此函数的结果,而无需重新运行即可映射多个scATAC-seq数据集。
首先,我们丢弃ATAC降维的第一个维度。
dims.atac <- 2:50
dims.rna <- 1:50
DefaultAssay(obj.multi) <- "RNA"
DefaultAssay(obj.rna) <- "SCT"
obj.rna.ext <- PrepareBridgeReference(
reference = obj.rna,
bridge = obj.multi,
reference.reduction = "spca",
reference.dims = dims.rna,
normalization.method = "SCT")
现在,我们可以直接在扩展参考和查询对象之间找到锚点。我们使用FindBridgeTransferAnchors函数,它使用与转换参考相同的字典来转换查询数据集,然后在该空间中识别锚点。该函数旨在模仿我们的FindTransferAnchors函数,但是要识别跨模态的对应关系。
bridge.anchor <- FindBridgeTransferAnchors(
extended.reference = obj.rna.ext,
query = obj.atac,
reduction = "lsiproject",
dims = dims.atac)
一旦我们识别了锚点,我们就可以将查询数据集映射到参考上。MapQuery函数与我们之前介绍的用于参考映射的函数相同。它从参考数据集中转移细胞注释,并且还在先前计算的UMAP嵌入上可视化查询数据集。由于我们的参考数据集包含三个分辨率级别的细胞类型注释(l1 - l3),我们可以将每个级别转移到查询数据集中。
obj.atac <- MapQuery(
anchorset = bridge.anchor,
reference = obj.rna.ext,
query = obj.atac,
refdata = list(
l1 = "celltype.l1",
l2 = "celltype.l2",
l3 = "celltype.l3"
),
reduction.model = "wnn.umap")
现在我们可以可视化结果,在参考UMAP嵌入上绘制基于其预测注释的scATAC-seq细胞。您可以看到,每个scATAC-seq细胞都根据scRNA-seq定义的细胞本体学被分配了一个细胞名称。
DimPlot(
obj.atac, group.by = "predicted.l2",
reduction = "ref.umap", label = TRUE
) + ggtitle("ATAC") + NoLegend()
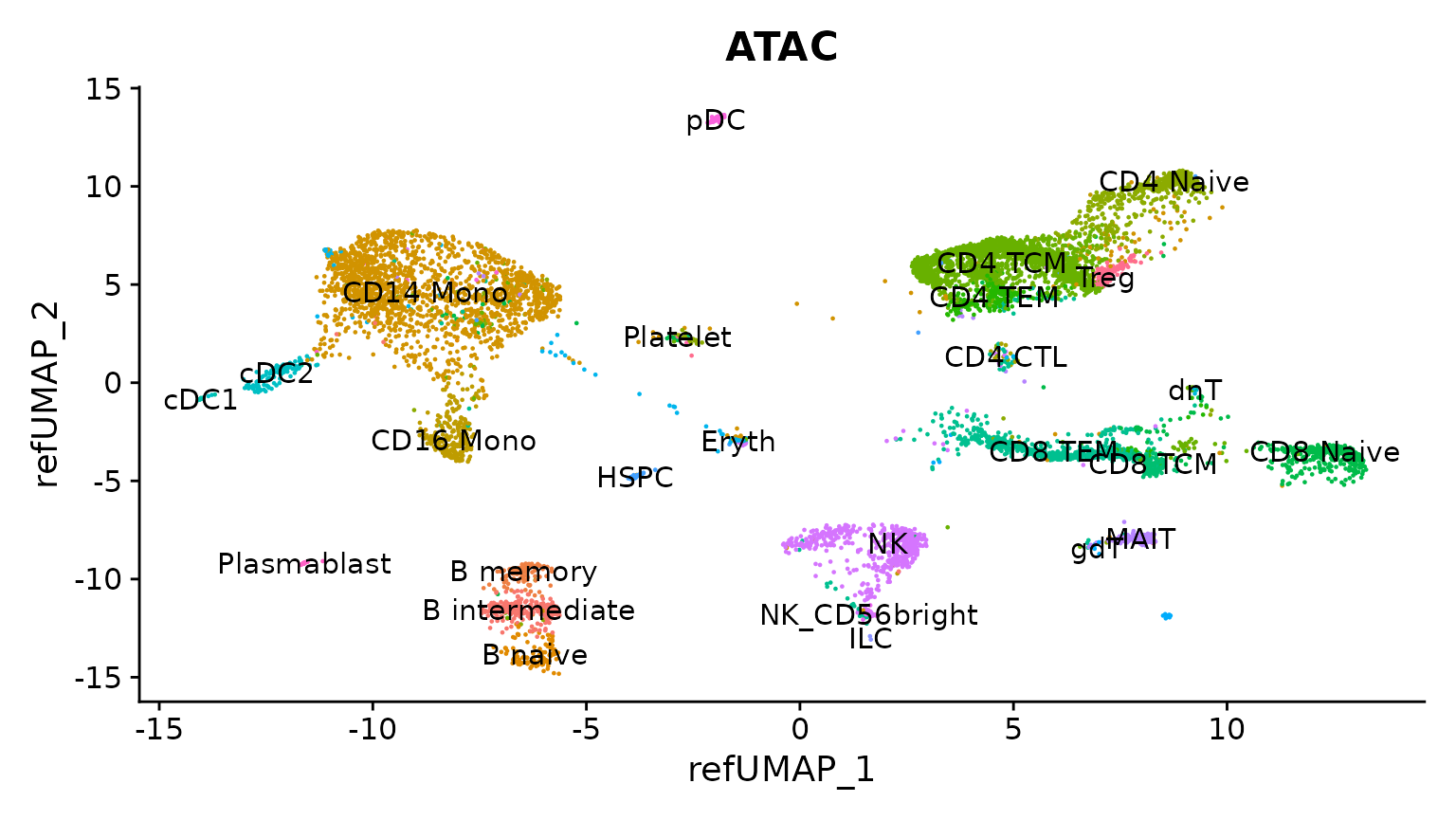
评估映射
为了评估映射和细胞类型预测,我们首先看看预测的细胞类型标签是否与scATAC-seq数据集的无监督分析一致。我们遵循scATAC-seq数据的标准无监督处理工作流程:
obj.atac <- FindTopFeatures(obj.atac, min.cutoff = "q0")
obj.atac <- RunSVD(obj.atac)
obj.atac <- RunUMAP(obj.atac, reduction = "lsi", dims = 2:50)
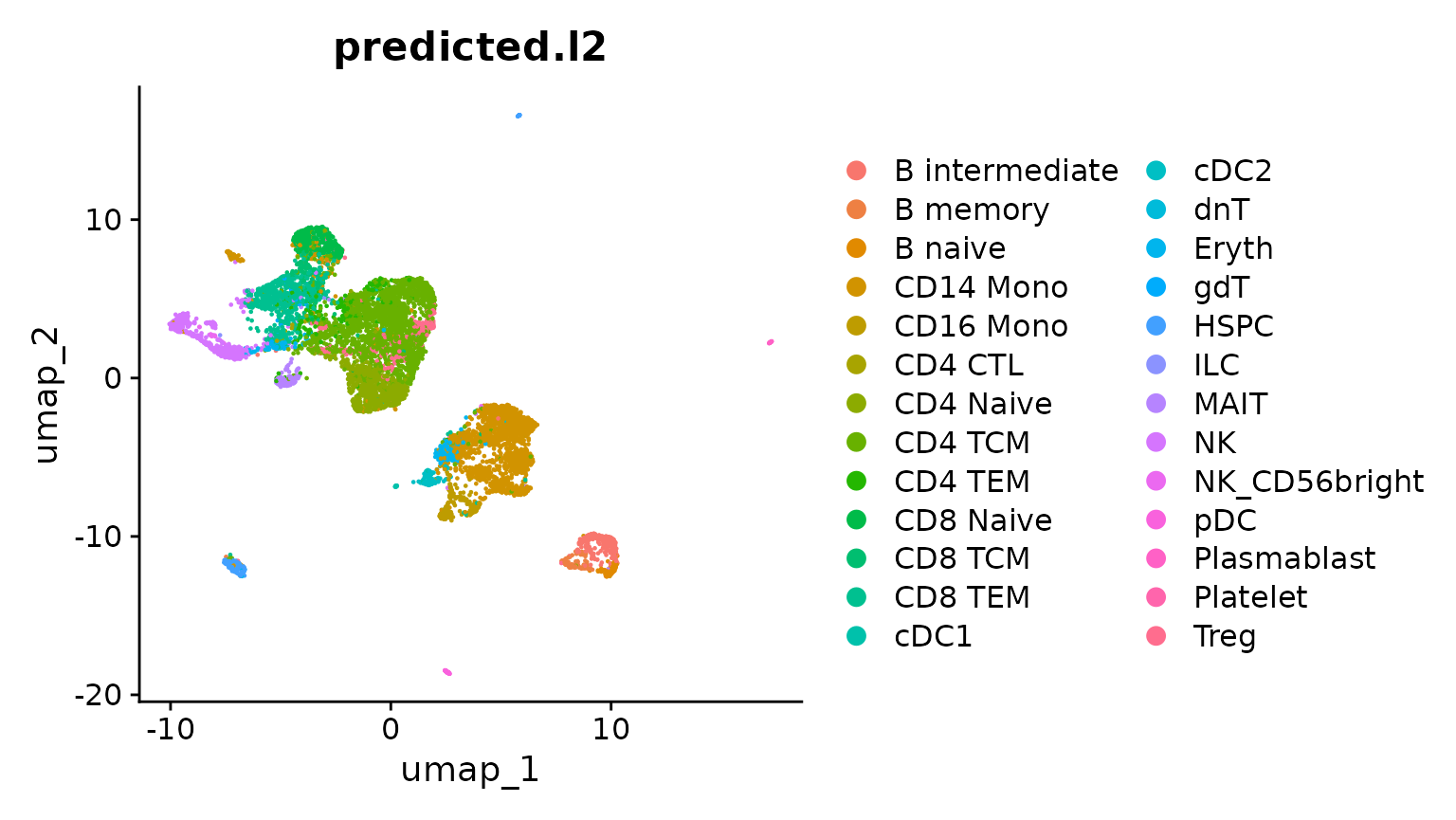
现在,我们在无监督UMAP嵌入上可视化预测的簇标签。我们可以看到,预测的簇标签(来自scRNA-seq参考)与scATAC-seq数据的结构一致。然而,有些细胞类型(例如Treg)在无监督分析中似乎没有分开。这些可能是预测错误,或者是参考映射提供额外分辨率的情况。
DimPlot(obj.atac, group.by = "predicted.l2", reduction = "umap", label = FALSE)
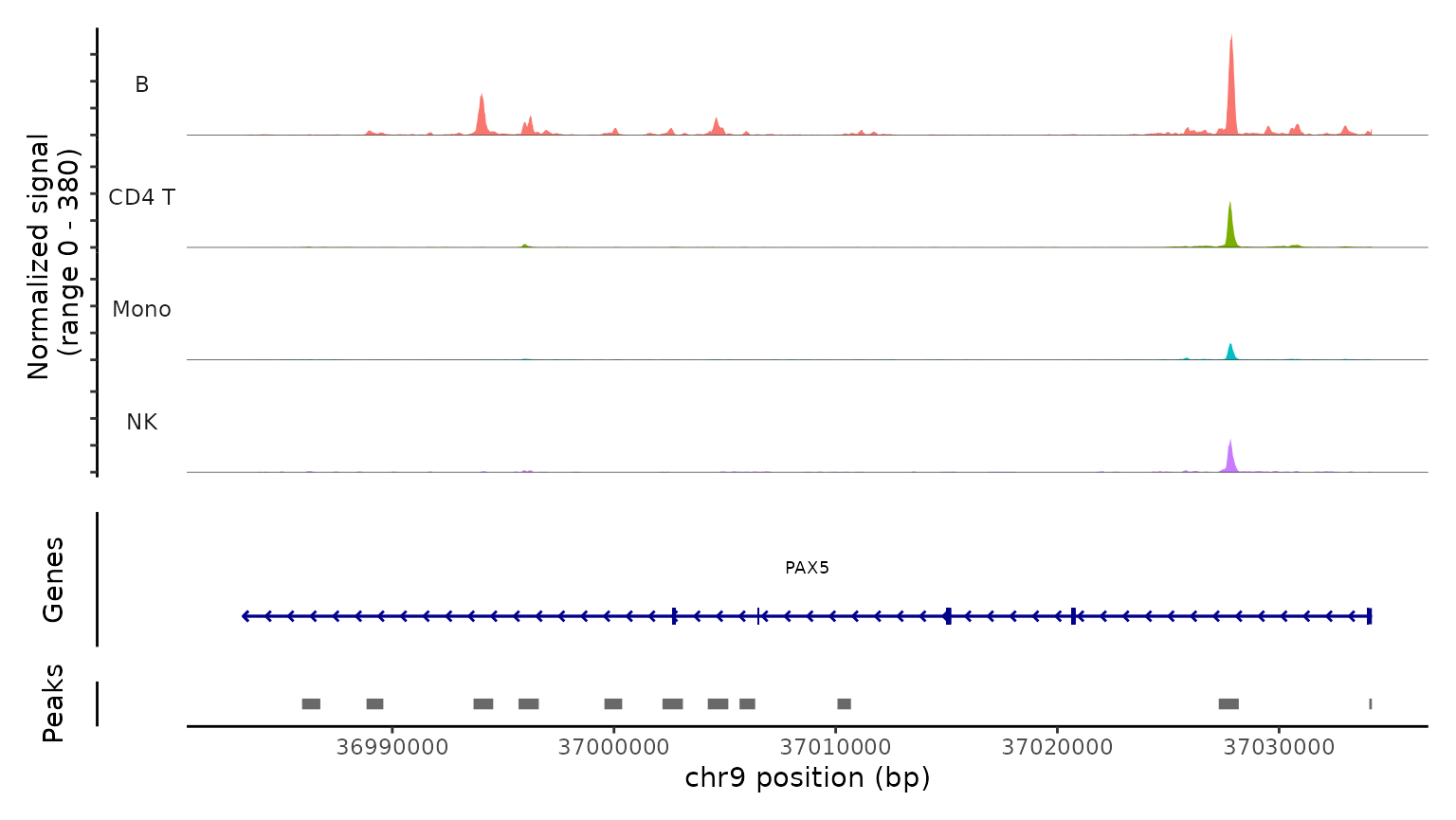
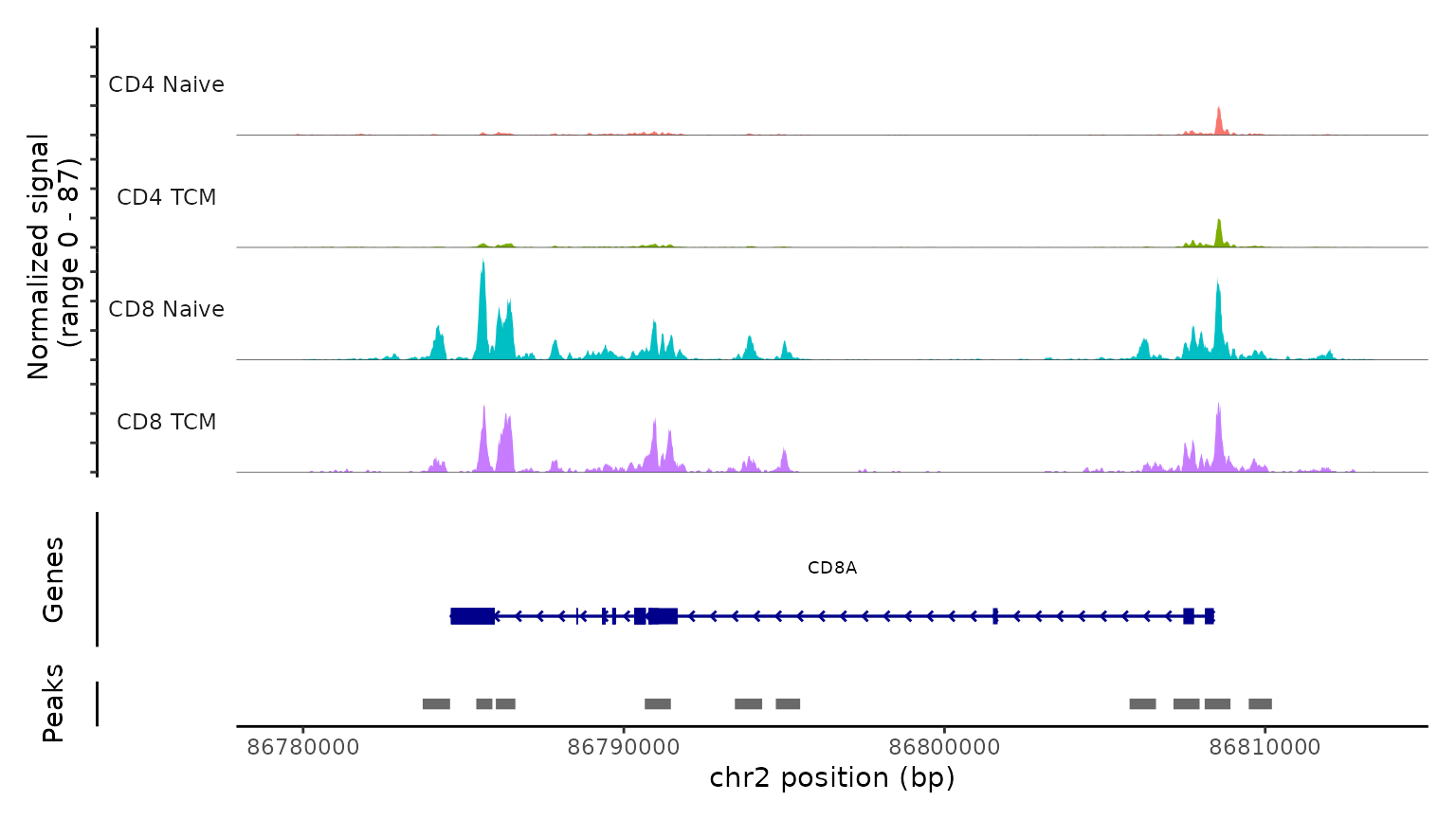
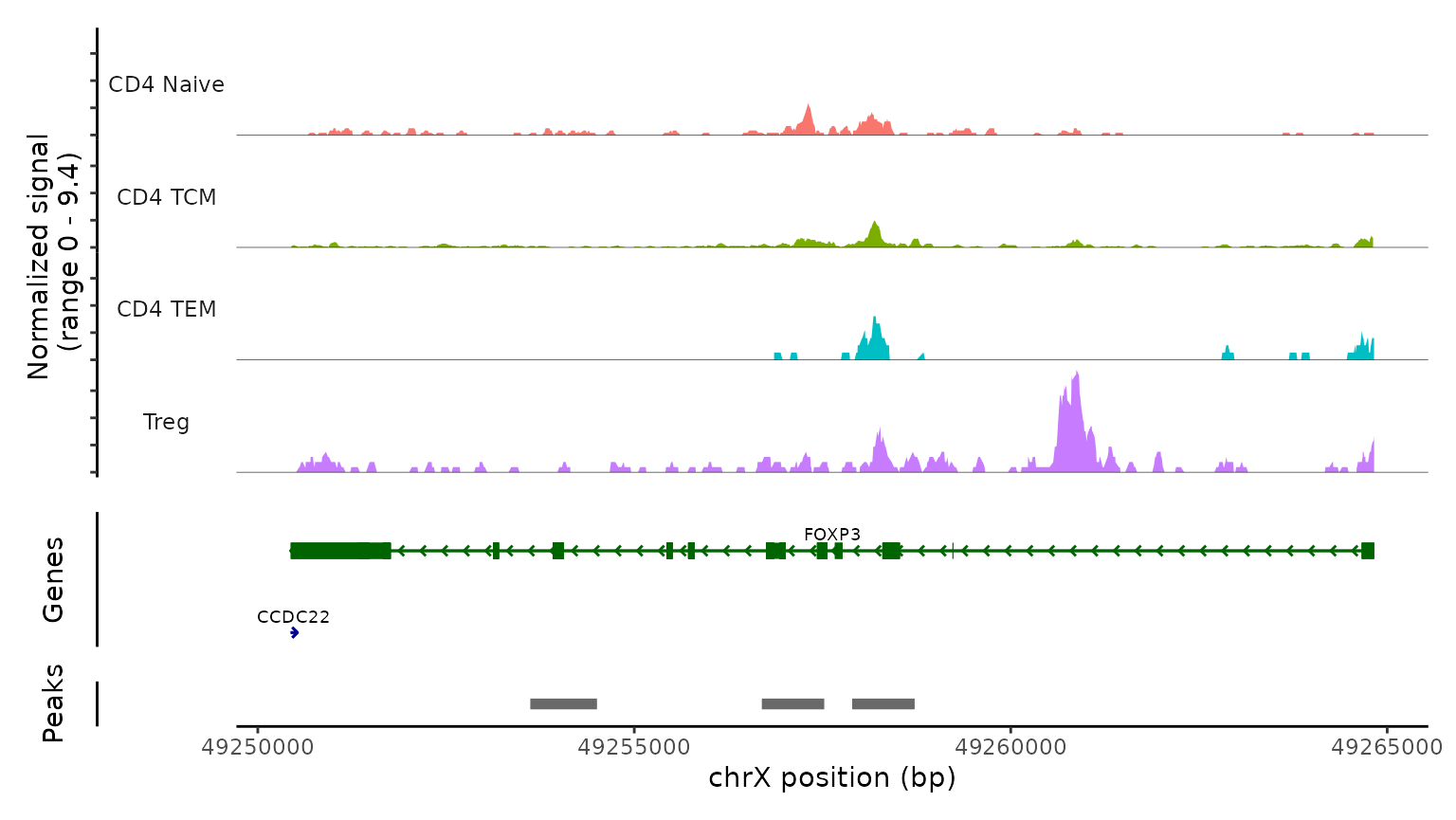
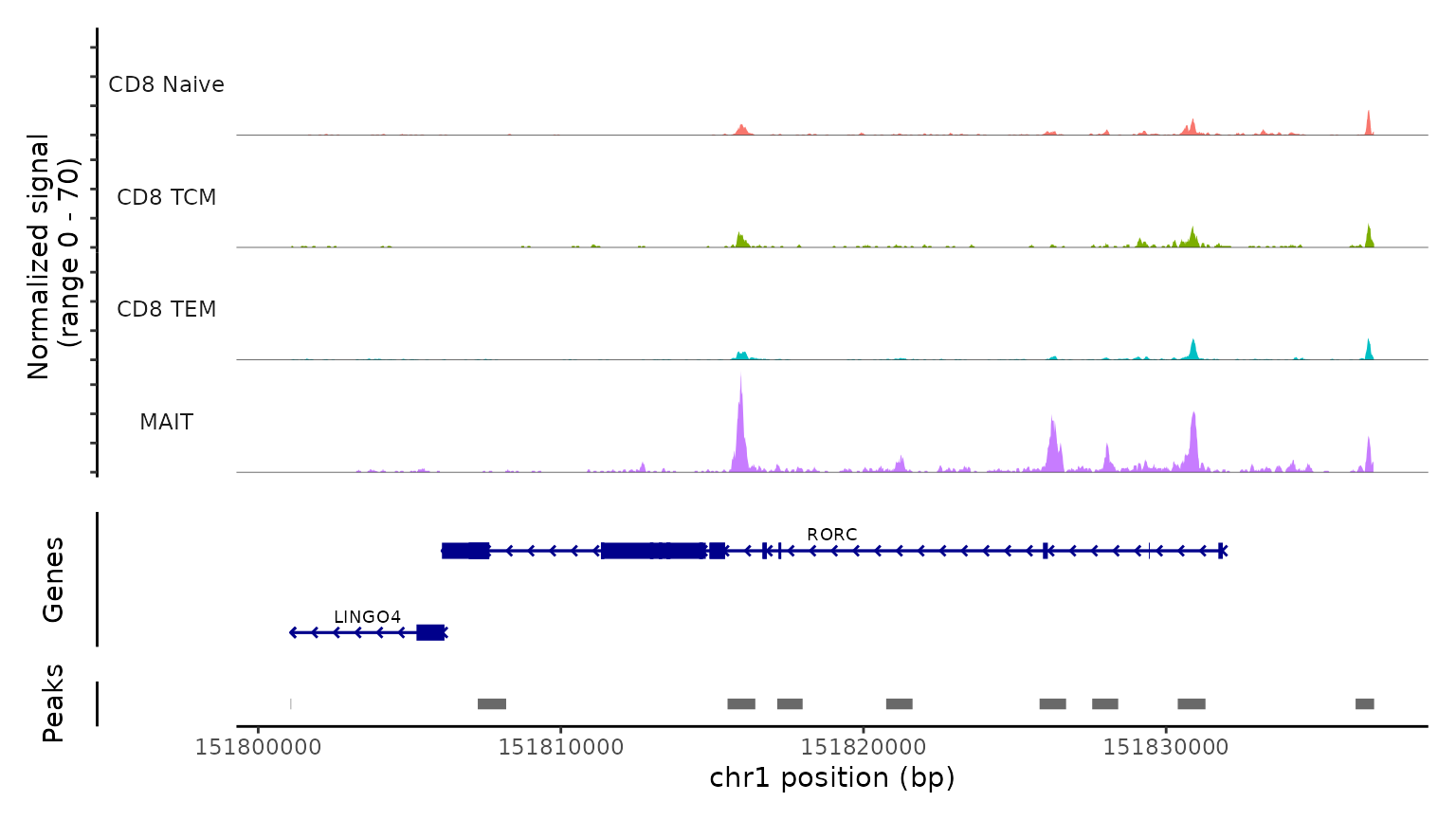
最后,我们通过检查scATAC-seq数据在典型位点的染色质可及性剖面来验证预测的细胞类型。我们使用CoveragePlot函数在CD8A、FOXP3和RORC处可视化可及性模式,按照其预测标签对细胞进行分组。我们在每种情况下都看到了预期的模式。例如,PAX5位点显示出仅在B细胞中可访问的峰,CD8A位点在CD8 T细胞亚群中也是如此。同样,预测的Tregs中FOXP3的可及性为我们的预测准确性提供了强有力的支持。
CoveragePlot(
obj.atac, region = "PAX5", group.by = "predicted.l1",
idents = c("B", "CD4 T", "Mono", "NK"), window = 200,
extend.upstream = -150000
)
CoveragePlot(
obj.atac, region = "CD8A", group.by = "predicted.l2",
idents = c("CD8 Naive", "CD4 Naive", "CD4 TCM", "CD8 TCM"),
extend.downstream = 5000, extend.upstream = 5000
)
CoveragePlot(
obj.atac, region = "FOXP3", group.by = "predicted.l2",
idents = c("CD4 Naive", "CD4 TCM", "CD4 TEM", "Treg"),
extend.downstream = 0, extend.upstream = 0
)
CoveragePlot(
obj.atac, region = "RORC", group.by = "predicted.l2",
idents = c("CD8 Naive", "CD8 TEM", "CD8 TCM", "MAIT"),
extend.downstream = 5000, extend.upstream = 5000
)
未完待续,欢迎关注!
本文由 mdnice 多平台发布















 677
677











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









