










文章目录
brief
这里主要根据seurat的教程走的,描述了多个单细胞数据集的整合,其中数据集的integration并不是简单的数据集的merge。
前者包括元信息的整合,数据集之间的批次矫正,后者仅仅是对数据表的拼接,后续直接renormalization即可。
同时这里描述的流程仅仅包括同类型的scRNA-seq测序数据,像scRNA-seq与scATAC-seq等多模态数据的整合暂未涉及。
此外像其他的单细胞数据集整合工具,例如harmony此处也没涉及。
library(dplyr)
library(Seurat)
library(patchwork)
library(sctransform)
library(ggplot2)
# devtools::install_github('satijalab/seurat-data')
library(SeuratData)
rm(list=ls())
# 获取测试数据集
# For convenience, we distribute this dataset through our SeuratData package.
# install dataset
InstallData("ifnb")
# load dataset
LoadData("ifnb")
Performing integration on datasets normalized with LogNormalize
# split the dataset into a list of two seurat objects (stim and CTRL)
ifnb.list <- SplitObject(ifnb, split.by = "stim")
# normalize and identify variable features for each dataset independently
ifnb.list <- lapply(X = ifnb.list, FUN = function(x) {
x <- NormalizeData(x)
x <- FindVariableFeatures(x, selection.method = "vst", nfeatures = 2000)
})
# select features that are repeatedly variable across datasets for integration <--repeatedly仅代表部分细胞在表达
# This function ranks features by the number of datasets they are deemed variable in, breaking ties by the median variable feature rank across datasets.
# It returns the top scoring features by this ranking.
features <- SelectIntegrationFeatures(object.list = ifnb.list)
immune.anchors <- FindIntegrationAnchors(object.list = ifnb.list, anchor.features = features)
# Returns a Seurat object with a new integrated Assay.
# this command creates an 'integrated' data assay
# If normalization.method = "LogNormalize", the integrated data is returned to the data slot and can be treated as log-normalized, corrected data.
# If normalization.method = "SCT", the integrated data is returned to the scale.data slot and can be treated as centered, corrected Pearson residuals
immune.combined <- IntegrateData(anchorset = immune.anchors)
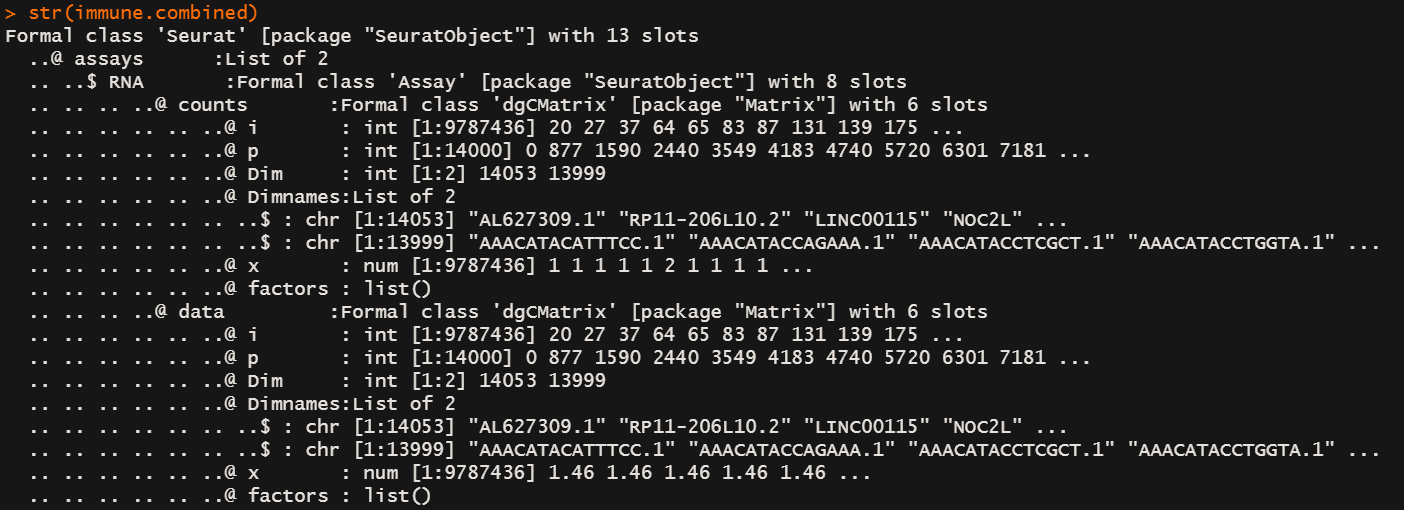
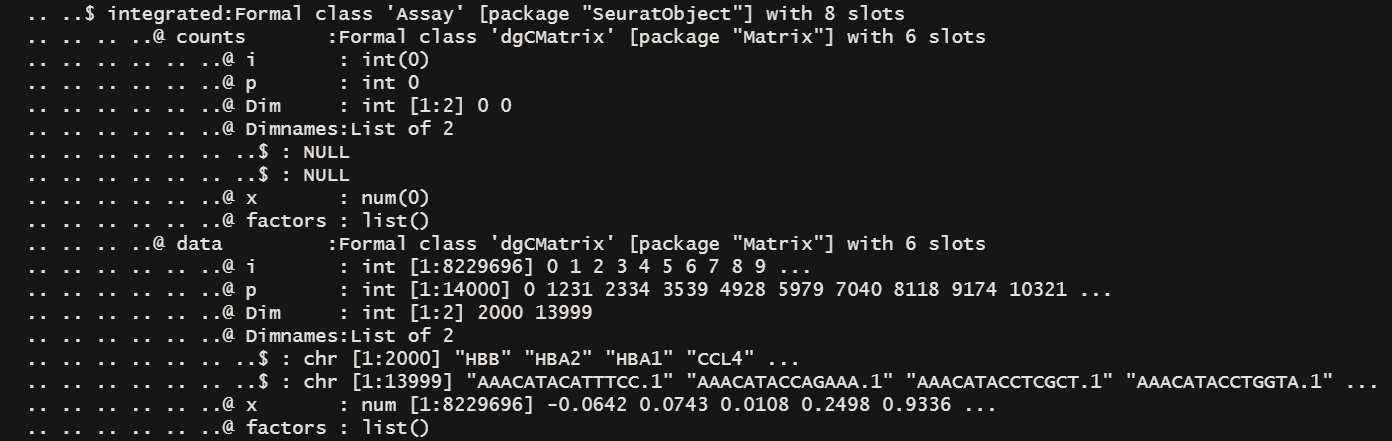
str(immune.combined) # 保留了2000个feature在Assays$integrated@data下面
# 确实看到了表达数据被修改了,至于是不是修正我是不敢说的
immune.combined@assays$integrated@data[immune.combined@assays$integrated@var.features,]
immune.combined@assays$RNA@data[immune.combined@assays$integrated@var.features,]
# specify that we will perform downstream analysis on the corrected data note that the
# original unmodified data still resides in the 'RNA' assay
DefaultAssay(immune.combined) <- "integrated"
# Run the standard workflow for visualization and clustering
immune.combined <- ScaleData(immune.combined, verbose = FALSE)
str(immune.combined) # 每个基因在所有细胞中进行了 cale
immune.combined <- RunPCA(immune.combined, npcs = 30, verbose = FALSE)
immune.combined <- RunUMAP(immune.combined, reduction = "pca", dims = 1:30)
immune.combined <- FindNeighbors(immune.combined, reduction = "pca", dims = 1:30)
immune.combined <- FindClusters(immune.combined, resolution = 0.5)
================================================================================
# Visualization
p1 <- DimPlot(immune.combined, reduction = "umap", group.by = "stim")
p2 <- DimPlot(immune.combined, reduction = "umap", label = TRUE, repel = TRUE)
p1 + p2
# To identify canonical cell type marker genes that are conserved across conditions, we provide the FindConservedMarkers() function.
# For performing differential expression after integration, we switch back to the original data
DefaultAssay(immune.combined) <- "RNA"
nk.markers <- FindConservedMarkers(immune.combined, ident.1 = 6, grouping.var = "stim", verbose = FALSE)
head(nk.markers)
-
整合前和整合后 anchors的数值变化
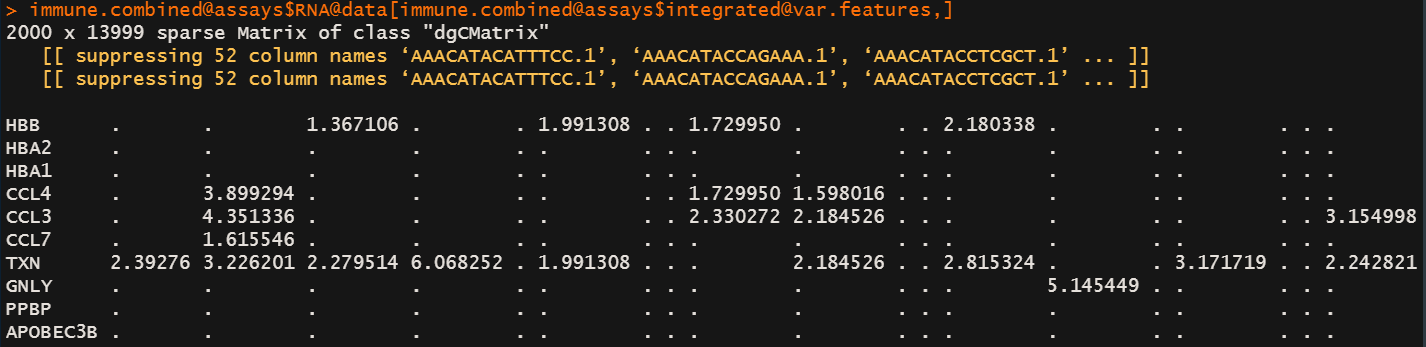

-
整合前的数据以及LogNormalization的数据一直存放在RNA@data@x下面
- 整合后的数据存放在integration@data@x
Performing integration on datasets normalized with SCTransform
# Performing integration on datasets normalized with SCTransform
# install glmGamPoi
if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager")
BiocManager::install("glmGamPoi")
# install sctransform from Github
install.packages("sctransform")
# load dataset
LoadData("ifnb")
# split the dataset into a list of two seurat objects (stim and CTRL)
ifnb.list <- SplitObject(ifnb, split.by = "stim")
# SCTransform只接受单个的seurat object
ctrl <- ifnb.list[["CTRL"]]
stim <- ifnb.list[["STIM"]]
ctrl.sct <- SCTransform(ctrl, vst.flavor = "v2", verbose = FALSE) %>%
RunPCA(npcs = 30, verbose = FALSE)
stim <- SCTransform(stim, vst.flavor = "v2", verbose = FALSE) %>%
RunPCA(npcs = 30, verbose = FALSE)
ifnb.list <- list(ctrl = ctrl, stim = stim)
# selecting a list of informative features using SelectIntegrationFeatures()
features <- SelectIntegrationFeatures(object.list = ifnb.list, nfeatures = 3000)
# To perform integration using the pearson residuals calculated above, we use the PrepSCTIntegration() function
ifnb.list <- PrepSCTIntegration(object.list = ifnb.list, anchor.features = features)
# To integrate the two datasets, we use the FindIntegrationAnchors() to find anchors
immune.anchors <- FindIntegrationAnchors(object.list = ifnb.list, normalization.method = "SCT",
anchor.features = features)
# and use these anchors to integrate the two datasets together with IntegrateData()
immune.combined.sct <- IntegrateData(anchorset = immune.anchors, normalization.method = "SCT")
# Perform an integrated analysis
immune.combined.sct <- RunPCA(immune.combined.sct, verbose = FALSE)
immune.combined.sct <- RunUMAP(immune.combined.sct, reduction = "pca", dims = 1:30, verbose = FALSE)
immune.combined.sct <- FindNeighbors(immune.combined.sct, reduction = "pca", dims = 1:30)
immune.combined.sct <- FindClusters(immune.combined.sct, resolution = 0.3)















 1万+
1万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









