






 本文介绍了如何使用DAVID工具进行GO和KEGG富集分析,包括上传基因列表、选择基因类型、指定物种和解析分析结果等步骤。通过对基因集合的功能富集,可以揭示基因集合的功能偏好性和生物学意义。
本文介绍了如何使用DAVID工具进行GO和KEGG富集分析,包括上传基因列表、选择基因类型、指定物种和解析分析结果等步骤。通过对基因集合的功能富集,可以揭示基因集合的功能偏好性和生物学意义。
富集分析相信大家都用的特别多,尤其在测序数据冗余的今天,不管是转录组、甲基化、ChIP-seq还是重测序,都会用到对一个或多个集合的基因进行功能富集分析。分析结果可以揭示这个集合的基因具有什么样的功能偏好性,进而可以对其相应的生物学意义做出判断。
今天给大家分享如何使用DAVID (the Database for Annotation,Visualization and Integrated Discovery),进行GO和KEGG富集分析。
(1)打开DAVID官网:https://david.ncifcrf.gov/
(2)点击左侧功能菜单:Functional Annotation

(3)进入到如下的页面中,页面中的大红框中就是进行分析所用的主要操作区域。进入分析页面后,通过如下三步即可完成分析:
提交基因列表 --> 选定提交列表类型 --> 开始分析
a、在 “Enter Gene List” 中上传基因列表,格式是每行一个基因。按照 DAVID 的要求,总的基因个数不得超过 3000 个。这里使用的基因序列:
AAK1
AIF1
ATP5J
BCL11B
CDC25B
CECR1
CENPT
CETN3
CHMP5
CISD2
CKLF
CLEC4D
CMTM2
TP53
MDM4
ATR
FZR1
CCND3
CCNE2
ATR
CKS1B
...
b、 “Select Identifier” 中选择上传的基因类型,因为我们上传的是基因名(Gene Symbol),所以在下拉菜单中选择 “OFFICIAL_GENE_SYMBOL”。
c、在 “List Type” 中有两个单选框,我们统一选择 “Gene List” 这一项。
d、点击 “Submit List” 即可

提交基因列表之后,经过几秒钟的等待,如果分析顺利,就会弹出一个提示:Please note that multiple species have been detected in your gene list. 这句话的意思就是在我们提交的基因列表中检测到多个物种,需要我们选择相应的物种。点击弹出框中的 “确定”,然后在 “List” 中的选择相应物种,于本次分析使用的是人类细胞,故这里我们选择 “Homo sapiens”,(读者可根据自己研究物种的类型进行选择)并点击下方的 “Select Species” 即可。
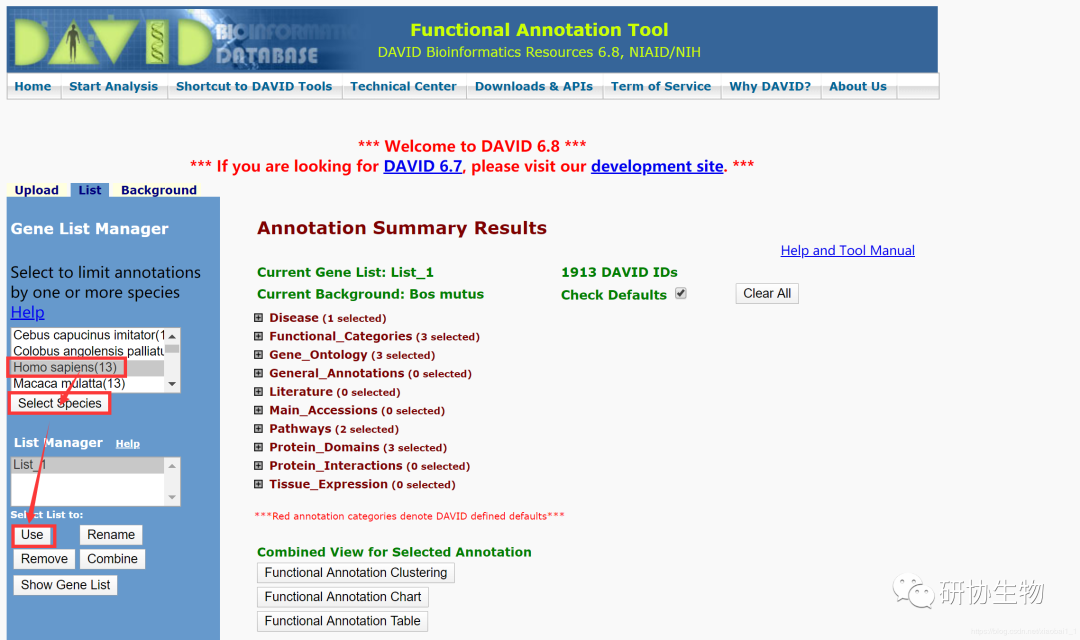
操作完成后,就可以得到如下图所示的分析结果。红框所示折叠框中分别就是GO和KEGG的分析结果。
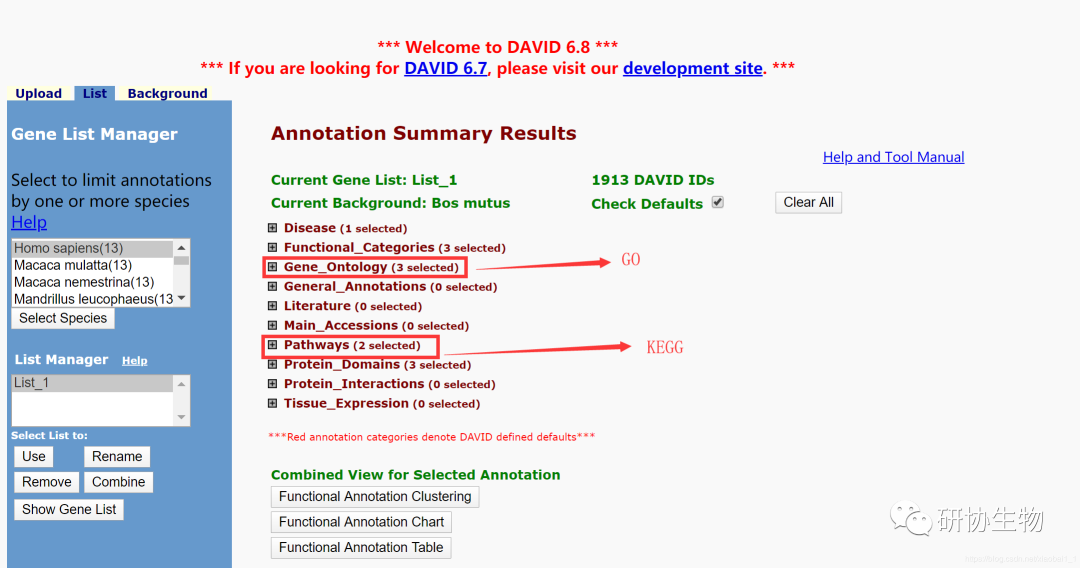
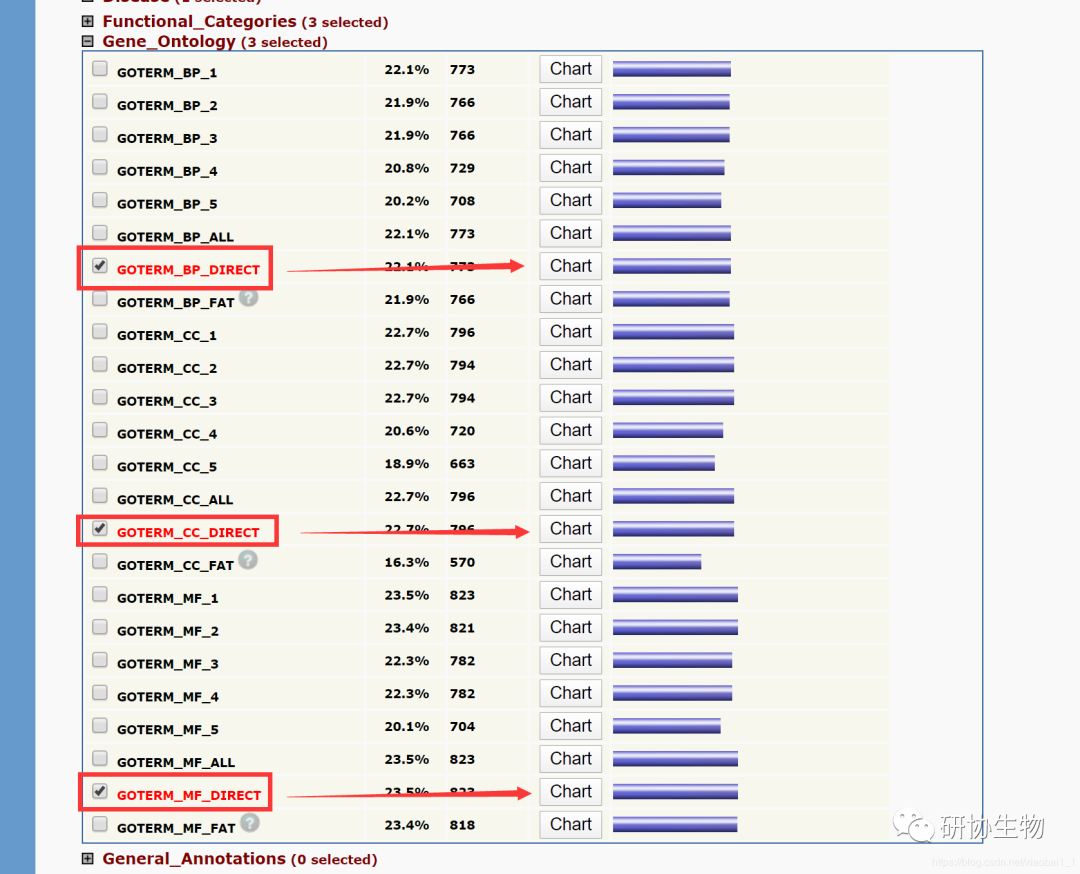
在功能富集分析的结果中有多个折叠栏,其中 Gene_Ontology 这一折叠栏中有有三个栏目:GOTERM_BP_FAT、GOTERM_CC_FAT、GOTERM_MF_FAT 就是我们想要的 GO 功能富集分析结果。而 Pathways 里面有一个 KEGG_PATHWAY 就是我们要的结果。找到 BP、CC、MF 和 KEGG 对应的详细结果, 点击每个栏目后面的 “Chart” 即可。
GO 富集分析
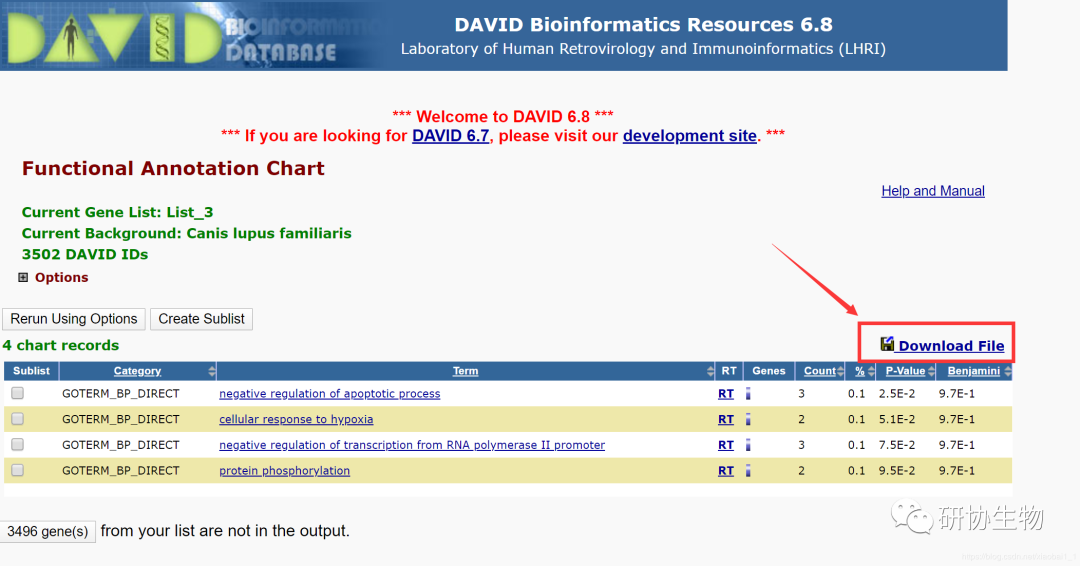
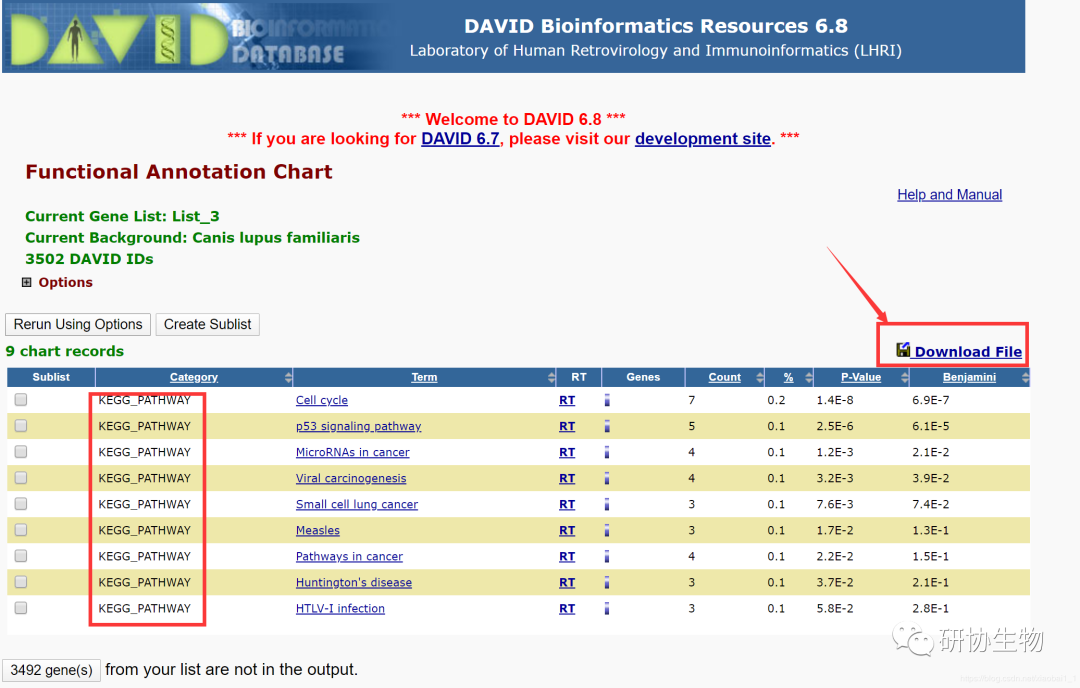
点击 “Chart” 之后,即可出现如下图所示的结果,这里面有几列数据分别是:Category、Term、RT、Genes、Count、%、P-Value 和 Benjamini。这几列中我们比较关心的是:Term(GO语义)、P-Value(P值)、Count(基因数)、%(基因比例)点击红框中的 Download File 即可。打开一个新的网页,新打开的网页就是分析结果的文本文件,可以下载或者导入到作图软件中进行后续的操作。
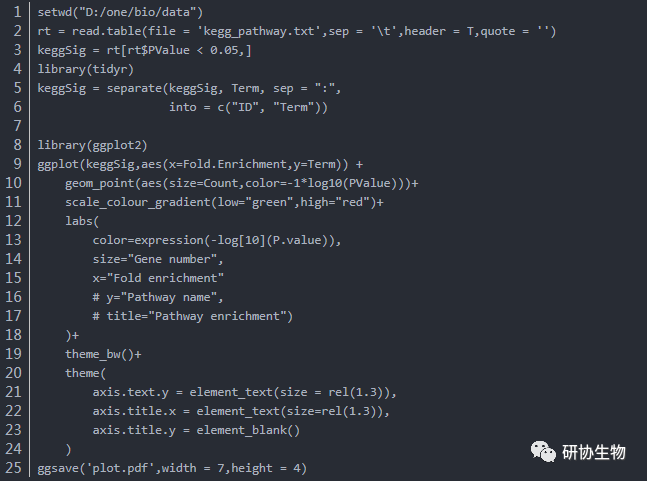
可视化富集分析结果复制所有富集分析结果,保存为“up_GO.txt”文件BP CC MF分别提取counts数前5的term:
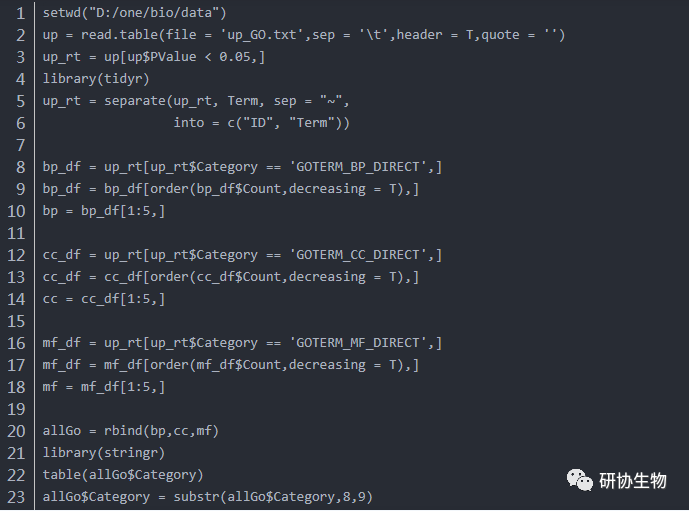
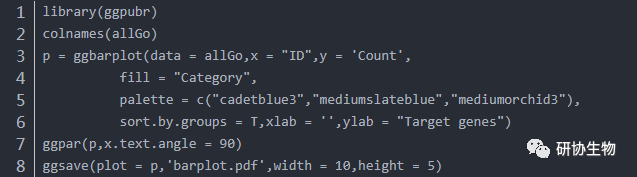
画出条形图:
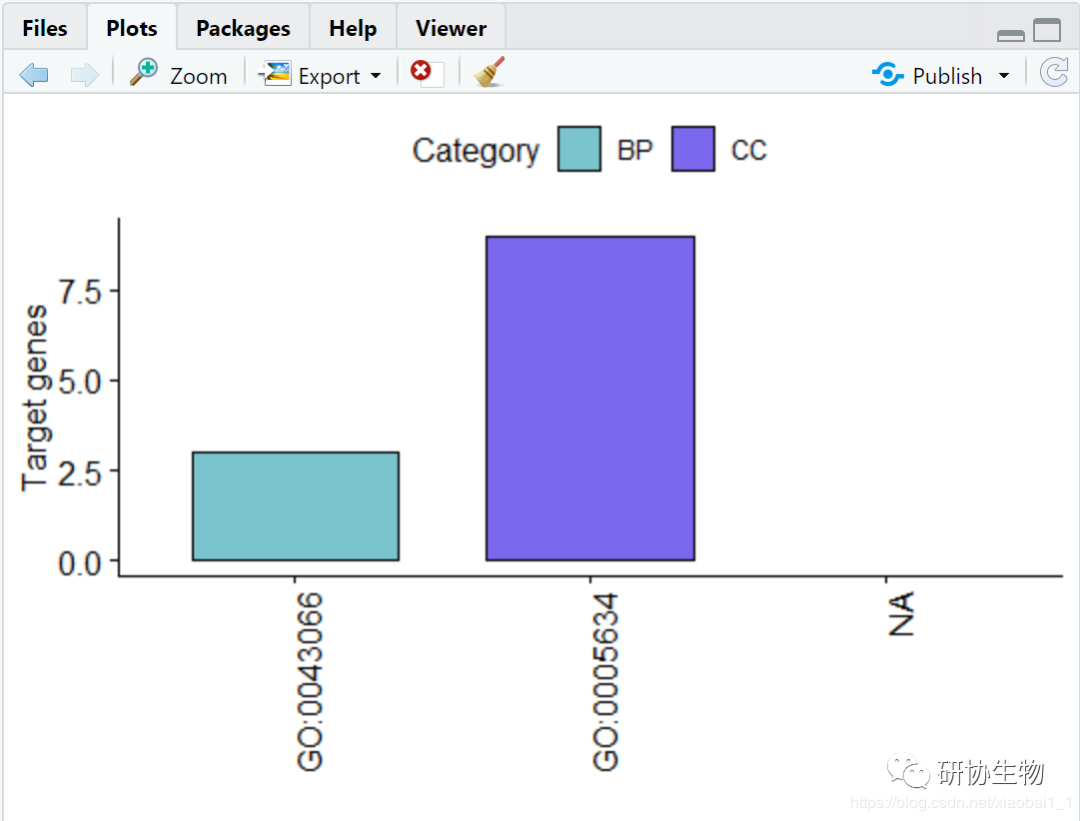
KEGG 富集分析
选择GO富集分析结果时,我们点击“Pathways (1 selected)”下拉选项,选择“KEGG_PATHWAY”选项。
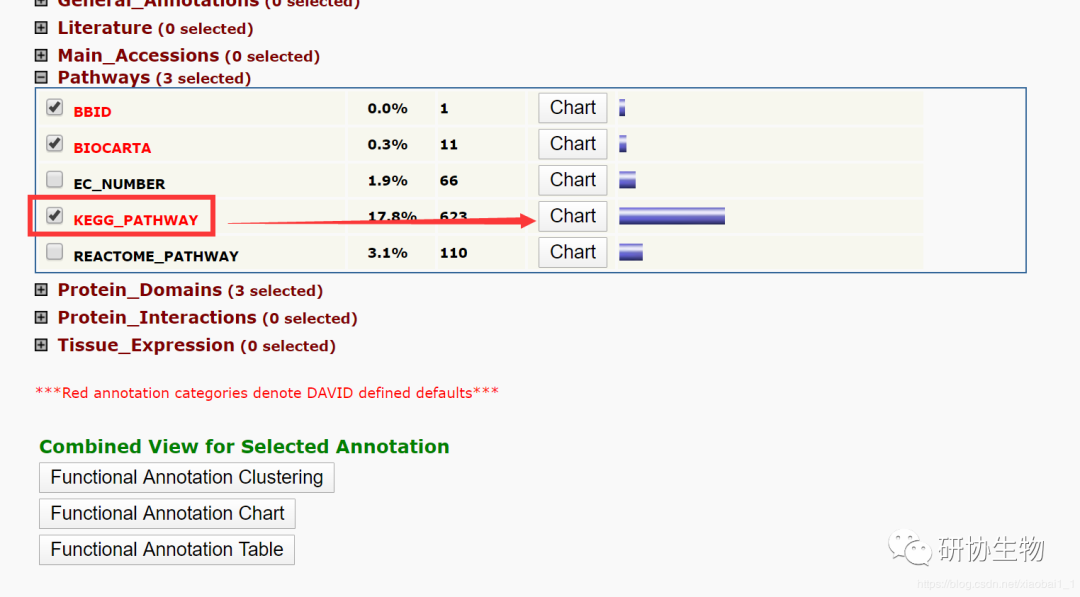
点击红框中的 Download File 即可保存文件kegg_pathway.txt
可视化富集分析结果(气泡图)
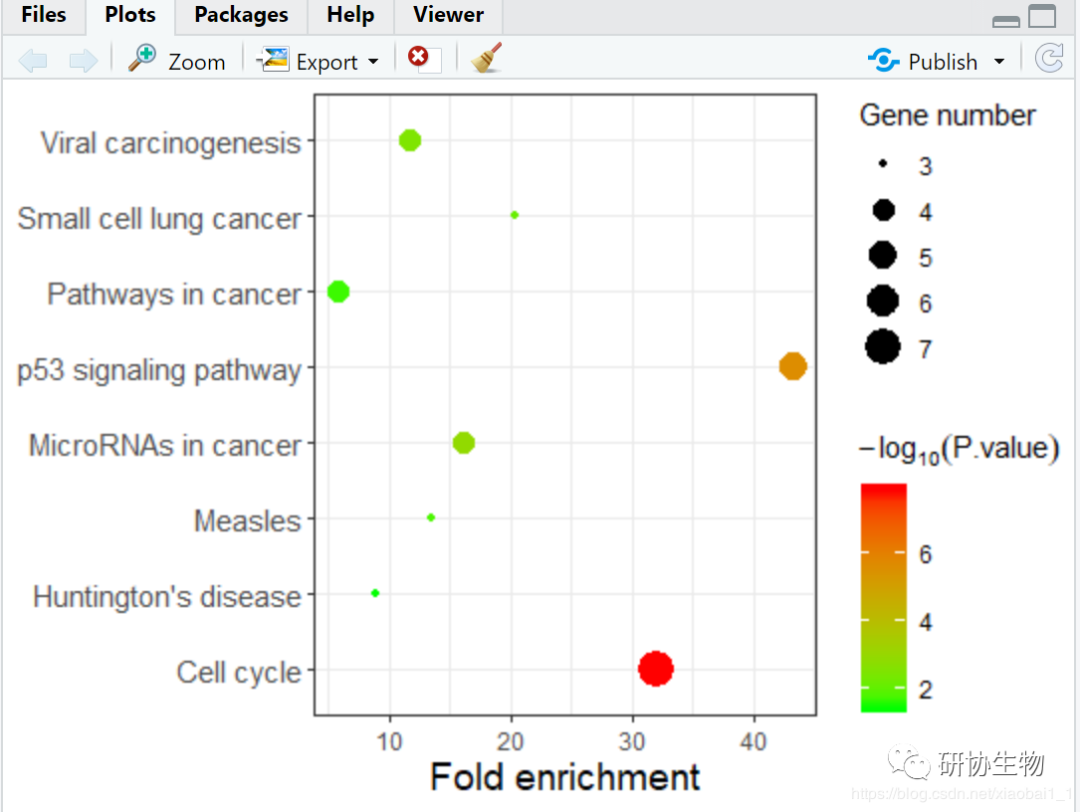
注:图中每一个点表示一个KEGG通路,通路名称见左侧坐标轴。横坐标为富集因子,表示差异表达蛋白中注释到该通路的蛋白比例与该物种蛋白注释到某通路的蛋白比例的比值。富集因子越大,表示差异蛋白在该通路中的富集显著性越可靠。
▼更多精彩推荐,请关注我们▼

















 3103
3103

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









