







之前分析的,和老师讨论之后,还是有问题。明天,要向唐老师汇报这件事(既然交给我,我就要把它认认真真的完成。)
##########################加载完成数据#############################################################
setwd("F://Rworkplace//唐老师的差异分析//结果文件整理")
library(dplyr)
library(Seurat)
library(patchwork)
library(ggplot2)
data<-read.csv("process.csv",header=T)
row.names(data)<-data[,1]
data<-data[,-1]
dim(data)
data_2 <- as(as.matrix(data),"dgCMatrix") #主要原因是没有加载包。
#rm(list=ls())
#gc()
#rstudioapi::restartSession()
# CreateSeuratObject
data_3<- CreateSeuratObject(counts = data_2, project = "tang", min.cells = 3, min.features = 200)
#最后创建secrut对象
########################################################################################
rm(data) #为了节省空间,删除原始矩阵
rm(data_2)
######################################################################################
#标准化,生成的标准化之后的矩阵存放在[["RNA"]]@data这个数据集中
data_3<-NormalizeData(data_3,normalization.method = "LogNormalize",scale.factor = 10000) #原来是我根本就没有对其进行标准化
data_3<-FindVariableFeatures(data_3,selection.method = "vst",nfeatures = 5633)
all.genes<-rownames(data_3)
data_3<-ScaleData(data_3,features = all.genes)
data_3<-RunPCA(data_3,features = VariableFeatures(object = data_3))normalize之后,遇到了问题,在我们normalize之后的矩阵中,有些变成了NA的缺失值。
所以,在PCA的过程中会出错。
Warning in irlba(A = t(x = object), nv = npcs, …) :
You’re computing too large a percentage of total singular values, use a standard svd instead.
Error in pca.results u u %*% diag(pca.results ud) :
non-conformable arguments
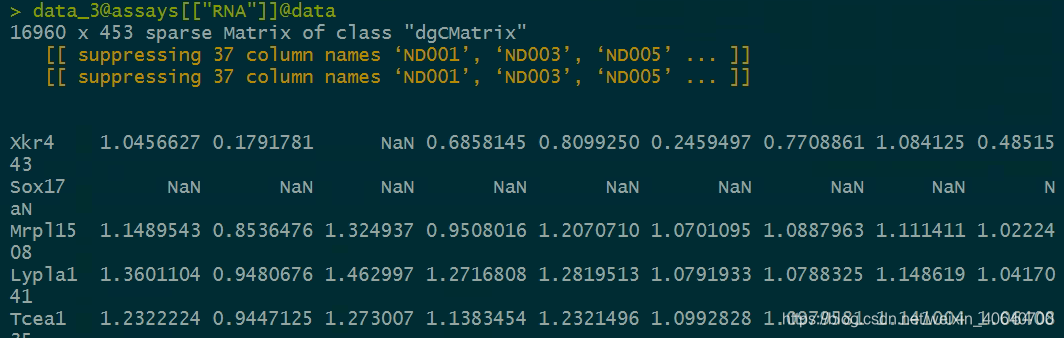
和我们的原始的count矩阵比较一下。
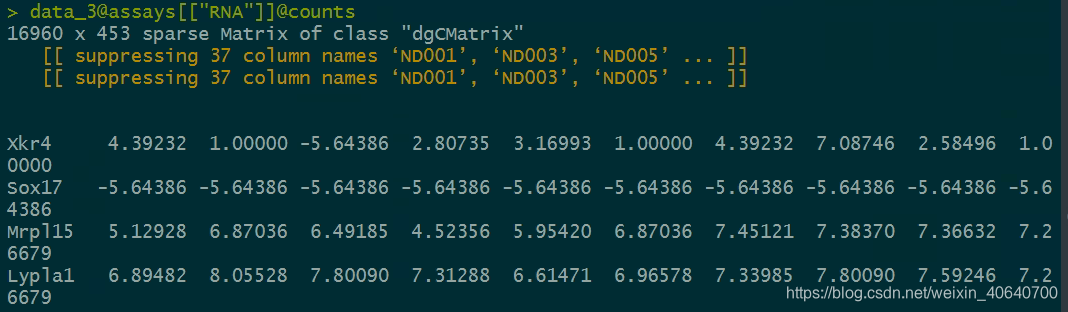 如果不进行normalize之后,会怎么样呢?
如果不进行normalize之后,会怎么样呢?
将normalize那行注释之后,重新开始跑。
##########################加载完成数据#############################################################
setwd("F://Rworkplace//唐老师的差异分析//结果文件整理")
library(dplyr)
library(Seurat)
library(patchwork)
library(ggplot2)
data<-read.csv("process.csv",header=T)
row.names(data)<-data[,1]
data<-data[,-1]
dim(data)
data_2 <- as(as.matrix(data),"dgCMatrix") #主要原因是没有加载包。
#rm(list=ls())
#gc()
#rstudioapi::restartSession()
# CreateSeuratObject
data_3<- CreateSeuratObject(counts = data_2, project = "tang", min.cells = 3, min.features = 200)
#最后创建secrut对象
########################################################################################
rm(data) #为了节省空间,删除原始矩阵
rm(data_2)
######################################################################################
#标准化,生成的标准化之后的矩阵存放在[["RNA"]]@data这个数据集中
#进行标准化之后,频繁出现的负值会被计算为NANs,从而在进行PCA的过程中出错。
#data_3<-NormalizeData(data_3,normalization.method = "LogNormalize",scale.factor = 10000) #原来是我根本就没有对其进行标准化
data_3<-FindVariableFeatures(data_3,selection.method = "vst",nfeatures = 5633)
all.genes<-rownames(data_3)
data_3<-ScaleData(data_3,features = all.genes)
data_3<-RunPCA(data_3,features = VariableFeatures(object = data_3))
然后进行聚类的分析。
data_3<-FindNeighbors(data_3,dims = 1:40)
data_3<-FindClusters(data_3,resolution = 1) #这个resolution的值是我试出来的……
(省略一些过程,最终的结果是)
t##########################加载完成数据#############################################################
sewd("F://Rworkplace//唐老师的差异分析//结果文件整理")
library(dplyr)
library(Seurat)
library(patchwork)
library(ggplot2)
data<-read.csv("process.csv",header=T)
row.names(data)<-data[,1]
data<-data[,-1]
dim(data)
data_2 <- as(as.matrix(data),"dgCMatrix") #主要原因是没有加载包。
#rm(list=ls())
#gc()
#rstudioapi::restartSession()
# CreateSeuratObject
data_3<- CreateSeuratObject(counts = data_2, project = "tang", min.cells = 3, min.features = 200)
#最后创建secrut对象
########################################################################################
rm(data) #为了节省空间,删除原始矩阵
rm(data_2)
######################################################################################
#标准化,生成的标准化之后的矩阵存放在[["RNA"]]@data这个数据集中
#data_3<-NormalizeData(data_3,normalization.method = "LogNormalize",scale.factor = 10000) #原来是我根本就没有对其进行标准化
data_3<-FindVariableFeatures(data_3,selection.method = "vst",nfeatures = 5633)
all.genes<-rownames(data_3)
data_3<-ScaleData(data_3,features = all.genes)
data_3<-RunPCA(data_3,features = VariableFeatures(object = data_3))
##########################################################################################
data_3<-FindNeighbors(data_3,dims = 1:40)
data_3<-FindClusters(data_3,resolution = 1) #这个resolution的值是我试出来的
##########################################################################################
data_3<-RunTSNE(data_3,dims = 1:40)
#data_3<-RunUMAP(data_3,dims = 1:36)
####################################################################################
data_3<-RenameIdents(data_3,
'0'='t1',
'1'='t2',
'2'='t3',
'3'='t4',
'4'='t5',
'5'='t6',
'6'='t7',
'7'='t8')
#################################################################################
DimPlot(data_3,reduction = "tsne",label = T)
#################################################################################
pdf("vinplot.pdf",11.69,28.27)
v1<-VlnPlot(data_3,features = c("Sprr1a"))
v2<-VlnPlot(data_3,features = c("Ecel1"))
v3<-VlnPlot(data_3,features = c("Sdc1"))
v4<-VlnPlot(data_3,features = c("Gpr151"))
v5<-VlnPlot(data_3,features = c("Srxn1"))
v6<-VlnPlot(data_3,features = c("Itga7"))
v7<-VlnPlot(data_3,features = c("Atf3"))
v8<-VlnPlot(data_3,features = c("Gal"))
v1+v2+v3+v4+v5+v6+v7+v8
dev.off()
#通过对图的观察,我们发现我们的结果和作者的结果还是有相通之处的。
#在这里会有些开心。
#################################################################################
筛选损伤的细胞。
########################################################################################
#在原始数据矩阵的基础上,筛去injured的细胞。
#筛去54个损伤的细胞
#细胞数量从453==>395
#筛选标准是sprr1a<10UMI,ecel1<10UMI
data<-read.csv("process.csv",header=T)
row.names(data)<-data[,1]
data<-data[,-1]
dim(data)
#36920 453
#首先将数据转换为secrut对象,我们这个矩阵就是count矩阵
#将数据转换为稀疏矩阵
#data_1<- as.matrix(data)
#在这里开始筛选和处理
#Sprr1a Ecel1
which(row.names(data)=="Sprr1a")
#3513
Sprr1a<-as.numeric(data[which(row.names(data)=="Sprr1a"),])
table(Sprr1a)
which(row.names(data)=="Ecel1")
#476
Ecel1<-as.numeric(data[which(row.names(data)=="Ecel1"),])
table(Ecel1)
#我现在想用知道这两者之间的交叉是什么?
#我先比较一下这两行
Sprr1a<-data[which(row.names(data)=="Sprr1a"),]
Ecel1<-data[which(row.names(data)=="Ecel1"),]
injury<-rbind(Sprr1a,Ecel1)
injury[3,]<-injury[1,]+injury[2,]
#table(injury[3,]>0)
#FALSE TRUE
# 392 61
#这个结果我很满意,那么接下来,我们就将TRUE的这些细胞筛去
#injury_cells <-colnames(injury[table(injury[3,]>0)])
#length(injury_cells)
which(injury[3,]>0)
#到这里不知道怎么做了,我想要提取这部分为true的细胞的名称
#Glp1r_cell<-colnames(processed_data[7324,which(processed_data[7324,]!="-5.64386")])
injury_cells<-colnames(injury[,which(injury[3,]>0)])
#成功了
uninjury_cells<-colnames(injury[,which(injury[3,]<=0)])
length(uninjury_cells)
data2<-subset(data,select = uninjury_cells)
dim(data2)
#这个data2的细胞和基因就是我们要处理得到的结果
write.csv(data2,"uninjury_cell.csv")
#####################################################################
#检验是否能够顺利的读取
data3<-read.csv("uninjury_cell.csv",header=T)
row.names(data3)<-data3[,1]
data3<-data3[,-1]
dim(data3)
#36920 392
#这个数据集就是我们想要的结果最后,就是真正的处理。
data<-read.csv("uninjury_cell.csv",header=T)
row.names(data)<-data[,1]
data<-data[,-1]
dim(data)
data_2 <- as(as.matrix(data),"dgCMatrix") #主要原因是没有加载包。
#rm(list=ls())
#gc()
#rstudioapi::restartSession()
# CreateSeuratObject
data_3<- CreateSeuratObject(counts = data_2, project = "tang", min.cells = 3, min.features = 200)
#最后创建secrut对象
########################################################################################
rm(data) #为了节省空间,删除原始矩阵
rm(data_2)
######################################################################################
#标准化,生成的标准化之后的矩阵存放在[["RNA"]]@data这个数据集中
#data_3<-NormalizeData(data_3,normalization.method = "LogNormalize",scale.factor = 10000) #原来是我根本就没有对其进行标准化
data_3<-FindVariableFeatures(data_3,selection.method = "vst",nfeatures = 3912)
all.genes<-rownames(data_3)
data_3<-ScaleData(data_3,features = all.genes)
data_3<-RunPCA(data_3,features = VariableFeatures(object = data_3))
##########################################################################################
data_3<-FindNeighbors(data_3,dims = 1:36)
data_3<-FindClusters(data_3,resolution = 2.8) #这个resolution的值是文献中的,我将3==>2.8,变成了12个cluster
#########################################################################################
data_3<-RunTSNE(data_3,dims = 1:36)
data_3<-RunUMAP(data_3,dims = 1:36)
#########################################################################################
data_3<-RenameIdents(data_3,
'0'='t1',
'1'='t2',
'2'='t3',
'3'='t4',
'4'='t5',
'5'='t6',
'6'='t7',
'7'='t8',
'8'='t9',
'9'='t10',
'10'='t11',
'11'='t12')
############################################################################
pdf("vinplot2.pdf",11.69,12.27)
v1<-VlnPlot(data_3,features = c("Gpr65"))
v2<-VlnPlot(data_3,features = c("Vip"))
v3<-VlnPlot(data_3,features = c("Glp1r"))
v4<-VlnPlot(data_3,features = c("Oxtr"))
v1+v2+v3+v4
dev.off()
############################################################################
DimPlot(data_3,reduction = "tsne",label = T)
############################################################################
#Oxtr Sst Calca vip Uts2b Edn3 Dbh Ebf3 Gpr65 Glp1r
pdf("tsne_genemarker.pdf",26.69,12.27)
p1<-FeaturePlot(data_3,features = c("Oxtr","Sst","Calca","Vip","Uts2b","Edn3","Dbh","Ebf3","Gpr65","Glp1r"),reduction = "tsne")
p1
dev.off()
############################################################################
#这个阈值设定的有些草莽,应该取3/4位数(3/4太小,使用90%,95%),作为cutoff值
data<-read.csv("process.csv",header=T)
row.names(data)<-data[,1]
data<-data[,-1]
dim(data)
which(row.names(data)=="Glp1r")
#18905
Glp1r_cell<-as.numeric(data[18905,])
quantile(Glp1r_cell,c(0.90))
#6.42626 95%
#5.75489 90%
which(row.names(data)=="Oxtr")
#7324
Oxtr_cell<-as.numeric(data[7324,])
quantile(Oxtr_cell,c(0.90))
#6.350086 95%
#4.57268 90%
which(row.names(data)=="Gpr65")
#15168
Gpr65_cell<-as.numeric(data[15168,])
quantile(Gpr65_cell,c(0.90))
#9.849222 95%
#9.383262 90%
which(row.names(data)=="Vip")
#12075
Vip_cell<-as.numeric(data[12075,])
quantile(Vip_cell,c(0.90))
#10.65393 95%
#9.025606 90%
############################################################################
cell1<-WhichCells(data_3,expression= Gpr65 > 9.4)
cell2<-WhichCells(data_3,expression= Vip > 9.0)
cell3<-WhichCells(data_3,expression= Glp1r > 5.8)
cell4<-WhichCells(data_3,expression= Oxtr> 4.6)
pdf("cluster_markecells.pdf",11.69,8.27)
plot1 <- DimPlot(data_3,label=T,cells.highlight= list(cell1), cols.highlight = c("blue"),cols = "grey",reduction = "tsne")+ggtitle("Gpr65 > 9.4(90%)")+theme(plot.title = element_text(hjust = 0.5))
plot2 <- DimPlot(data_3,label=T,cells.highlight= list(cell2), cols.highlight = c("blue"),cols = "grey",reduction = "tsne")+ggtitle("Vip > 9.0(90%)")+theme(plot.title = element_text(hjust = 0.5))
plot3 <- DimPlot(data_3,label=T,cells.highlight= list(cell3), cols.highlight = c("blue"),cols = "grey",reduction = "tsne")+ggtitle("Glp1r > 5.8(90%)")+theme(plot.title = element_text(hjust = 0.5))
plot4 <- DimPlot(data_3,label=T,cells.highlight= list(cell4), cols.highlight = c("blue"),cols = "grey",reduction = "tsne")+ggtitle("Oxtr> 4.6(90%)")+theme(plot.title = element_text(hjust = 0.5))
plot1+plot2+plot3+plot4
dev.off()
###########################################################################
#确定要比较的类是t1-c(t2,t4,t10,t11)
cluster.markers<-FindMarkers(data_3,ident.1 = "t1",ident.2 =c("t2","t4","t10","t11") )
class(cluster.markers)
write.csv(cluster.markers,"deg.csv")
head(cluster.markers,n=5)
###########################################################################
pdf("tsne_deg_1.pdf",12.69,12.27)
p1<-FeaturePlot(data_3,features = c("Tmie","Scn5a","Ctxn2","Zbtb7c","Rprml","Rnf112"),reduction = "tsne")
p1
dev.off()
###########################################################################
pdf("vinplot3.pdf",11.69,18.27)
v1<-VlnPlot(data_3,features = c("Tmie"))
v2<-VlnPlot(data_3,features = c("Scn5a"))
v3<-VlnPlot(data_3,features = c("Ctxn2"))
v4<-VlnPlot(data_3,features = c("Zbtb7c"))
v5<-VlnPlot(data_3,features = c("Rprml"))
v6<-VlnPlot(data_3,features = c("Rnf112"))
v1+v2+v3+v4+v5+v6
dev.off()
#########################################################
#Kcnk18 Ntsr1 Olfm3 Greb1 Dcdc2a Chrna6
pdf("vinplot4.pdf",11.69,18.27)
v1<-VlnPlot(data_3,features = c("Kcnk18"))
v2<-VlnPlot(data_3,features = c("Ntsr1"))
v3<-VlnPlot(data_3,features = c("Olfm3"))
v4<-VlnPlot(data_3,features = c("Greb1"))
v5<-VlnPlot(data_3,features = c("Dcdc2a"))
v6<-VlnPlot(data_3,features = c("Chrna6"))
v1+v2+v3+v4+v5+v6
dev.off()
#####################################################################
#end
最终,完成我们的需求。
现在还剩下一个关键性的问题:
为什么?-5.64386这个值在进行normalize处理之后会变成NaN?
- 首先,需要明白,在R中,经过怎样的处理,才可能变成NaN,也就是NaN是什么意思?
R中的无定义数用NaN表示,即“Not a Number(非数)”
那,什么情况下,会出现这样的一个结果呢?
0/0
[1] NaN
也就是说,当我们的运算不符合数学规则的时候,通常会返回这样的一个值。这里的数学规则,如上面所示的,被除数不能为零。还有这下面的log()的定义域是大于零的,所以这时候-1就是不合法的。
log(-1)
[1] NaN
Warning message:
In log(-1) : NaNs produced
- 那看一下我们的数据,我们做了怎样的运算,使之产生了这样的结果。
首先,我想问一下NormalizeData如何运算的?
LogNormalize:
Feature counts for each cell are divided by the total counts for that cell and multiplied by the scale.factor.
This is then natural-log transformed using log1p
参考链接:https://www.rdocumentation.org/packages/Seurat/versions/4.0.3/topics/NormalizeData
找到问题的原因了:
> sum(data_3@assays[["RNA"]]@counts[,1])
[1] 23802.96 #问题的症结在这里,我这边的count的和是一个正数,所以-5.64386在最后运算的时候,就会出错。
> data_3@assays[["RNA"]]@counts[1,1]
[1] 4.39232
> data_3@assays[["RNA"]]@counts[2,1]
[1] -5.64386
> (data_3@assays[["RNA"]]@counts[2,1]/sum(data_3@assays[["RNA"]]@counts[,1]))*10000
[1] -2.371075
> a<-(data_3@assays[["RNA"]]@counts[2,1]/sum(data_3@assays[["RNA"]]@counts[,1]))*10000
> log(a)
[1] NaN
Warning message:
In log(a) : NaNs produced
但是啊但是,我们得到的这个count矩阵,是我们前面筛选之后的结果,我们原始的那个数据矩阵就没有这种情况。
> dim(data)
[1] 36920 453
> dim(data_3@assays[["RNA"]]@counts)
[1] 16960 453从这个数据集上看,整整筛了一大半。而具体的筛的过程是这样的。
data_3<- CreateSeuratObject(counts = data_2, project = "tang", min.cells = 3, min.features = 200)min.features
Include features detected in at least this many cells.
Will subset the counts matrix as well.
To reintroduce excluded features, create a new object with a lower cutoff.
min.cells
clude cells where at least this many features are detected.
通过上面的比较,cell的数量没有什么变化,而feature的数量却大大减少。从36920==>16960。
因为我们一共有453个细胞,而将其设定为200,确实有点狠。
而且,她这边筛选的标准,我觉得应该更像是对于0值而言的,但是我们的数据中的0,不是这个0,所以,从某种程度上讲,还是有问题。如果让我去修改的话,应该是
data_3<- CreateSeuratObject(counts = data_2, project = "tang", min.cells = 0, min.features = 0)我觉得这样,应该更加合理一些。我决定换用这个重新处理。
之前,之所以会犯错,我觉得是我对这个流程了解的太少了。只有更进一步的了解,才能够调整它每一步的细节。
不过目前感到比较欣慰的是,作者在文章中提到的这些基因,居然不会因为上述过程被筛去。我能够继续在这个基础上,画图,也是神奇。对于这个数据很迷。
可能本身也没有那么多的零吧。我瞅瞅我之前统计的。
-5.64386 11617595
0 190512
1 129560
#我低估了,0和1是仅次于-5.64386的值,只是为什么他们也这么多,以及和-5.64386的关系,我暂时想不太明白。
下午的汇报,我不知道自己是否能够自圆其说。有点忐忑,害怕不专业。
不过这篇文章,在这里就结束了。

















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









